Abstracts
The main objective of the present study was to determine the permeability of clarithromycin (CLA)-PLGA nanoparticles using single-pass intestinal perfusion technique in rats. Clarithromycin nanoparticles were prepared by nano-precipitation according to the modified quasi emulsion solvent diffusion technique and evaluated for their physicochemical characteristics. Permeability coefficients (Peff) in anaesthetized rats were determined at 3 different concentrations. Drug solution or suspensions in PBS was perfused through a cannulated jejunal segment and samples were taken from outlet tubing at different time points up to 90 min. Microbiological assay of CLA and phenol red in the samples were analyzed using an agar well diffusion procedure and HPLC method respectively. The average particle size of prepared nanoparticles was 305 ± 134 nm. The mean Peff of CLA solution in concentrations of 150, 250 and 400 µg/mL was found to be 1.20 (±0.32) ×10-3, 9.62 (±0.46) ×10-4, and 1.36 (±0.95) ×10-3 cm/sec, respectively. The corresponding values for the same concentration of nanoparticles were found to be 2.74 (±0.73) ×10-3, 2.45 (±0.88) ×10-3, and 3.68 (±0.46) ×10-3 cm/s, respectively. The two-tailed Student’s t-test showed that the intestinal permeability of CLA nanoparticle suspensions in prepared concentrations were significantly increased in comparison with its solution.
Clarithromycin/nanoparticles/ permeability; Single-Pass Intestinal Perfusion; Intestinal permeability
O objetivo principal do presente estudo foi determinar a permeabilidade de nanopartículas de claritromicina (CLA)-PLGA, utilizando a técnica de perfusão intestinal de passo único em ratos. As nanopartículas de claritromicina foram preparadas por nanoprecipitação, de acordo com a técnica modificada de difusão de solvente quase-emulsão, e suas características físico-químicas avaliadas. Os coeficientes de permeabilidade (Peff) em ratos anestesiados foram determinados em três concentrações diferentes. A solução, ou suspensões, do fármaco em PBS foi perfundida através do segmento de jejuno canulado e as amostras foram tomadas do tubo externo em diferentes tempos até 90 minutos. Os ensaios microbiológico de CLA e de vermelho de fenol das amostras foram realizados, utilizando-se o procedimento de difusão em poço de ágar e de CLAE, respectivamente. O tamanho médio das partículas das nanopartículas preparadas foi de 305 ± 134 nm. O Peff médio da solução de CLA em concentrações de 150, 250 and 400 µg/mL foi de 1.20(±0.32)×10-3, 9.62(±0.46)]×10-4 e de 1.36(±0.95)×10-3 cm/s, respectivamente. O valor correspondente para a mesma concentração de nanopartículas foi de 2.74 (±0.73)×10-3, 2.45(±0.88)×10-3 e de 3.68 (±0.46)×10-3 cm/s, respectivamente. O teste t de Student com duas variáveis mostrou que a permeabilidade intestinal das suspensões de nanopartículas de CLA nas concentrações preparadas foram significativamente aumentadas em comparação com sua solução.
Claritromicina/nanopartículas/permeabilidade; Perfusão Intestinal de Único Passo; Permeabilidade intestinal
INTRODUCTION
Oral administration remains the most convenient and useful route for delivering most pharmaceutical agents. However, the major problem of many orally administered drugs is to overcome several barriers before reaching their target site (Cook, Shenoy, 2003COOK, T.J.; SHENOY, S.S. Intestinal permeability of chlorpyrifos using the single-pass intestinal perfusion method in the rat. Toxicology, v.184, p.125-133, 2003.; Rao et al., 2008RAO, S.V.; YAJURVEDI, K.; SHAO, J. Self-nanoemulsifying drug delivery system (SNEDDS) for oral delivery of protein drugs: III. In vivo oral absorption study. Int. J. Pharm., v.362, p.16-19, 2008.). Several approaches have been applied in order to improve the oral bioavailability of poorly permeable and soluble compounds intended for oral administration. Using nanoparticulate drug delivery system is considered as one of these strategies (Cai et al., 2010COOK, T.J.; SHENOY, S.S. Intestinal permeability of chlorpyrifos using the single-pass intestinal perfusion method in the rat. Toxicology, v.184, p.125-133, 2003.; Saha et al., 2010SAHA, R.N.; VASANTHAKUMAR, S.; BENDE, G.; SNEHALATHA, M. Nanoparticulate drug delivery systems for cancer chemotherapy. Mol. Membr. Biol., v.27, p.215-231, 2010.). These formulations have been shown to be efficient approaches to enhance the transport of a large number of drugs including nucleic acids and genes across many biological membranes as well as to improve the stability of these materials. This could result in enhanced oral bioavailability of poorly bioavailable drugs due to their specialized uptake mechanisms such as circumventing the P-gp efflux and protecting incorporated drug molecules from the gastro-intestinal tract (GIT) degradation as well as gut wall and first pass metabolism (Arayne, Sultana, 2006ARAYNE, M.S.; SULTANA, N. Review: nanoparticles in drug delivery for the treatment of cancer. Pak. J. Pharm. Sci., v.19, p.258-268, 2006.; Florence, 2004FLORENCE, A.T. Issues in oral nanoparticle drug carrier uptake and targeting. J. Drug Target., v.12, p.65-70, 2004.; Moinard-Checot et al., 2006MOINARD-CHECOT, D.; CHEVALIER, Y.; BRIANCON, S.; FESSI, H.; GUINEBRETIERE, S. Nanoparticles for drug delivery: review of the formulation and process difficulties illustrated by the emulsion-diffusion process. J. Nanosci. Nanotechnol., v.6, p.2664-2681, 2006.). Since the intestinal epithelium is one of the main obstacles the drugs should pass through, it is both a great interest and a medical need for improving the intestinal permeability of various poorly bioavailable drugs. Intestinal permeability or the ability of a compound to move across the intestinal epithelial barrier is an important and critical determinant of its rate and extent of absorption (Zakeri-Milani et al., 2009aZAKERI-MILANI, P.; BARZEGAR-JALALI, M.; AZIMI, M.; VALIZADEH, H. Biopharmaceutical classification of drugs using intrinsic dissolution rate (IDR) and rat intestinal permeability. Eur. J. Pharm. Biopharm., v.73, p.102-106, 2009a.; Zakeri-Milani et al., 2007ZAKERI-MILANI, P.; VALIZADEH, H.; TAJERZADEH, H.; AZARMI, Y.; ISLAMBOLCHILAR, Z.; BARZEGAR, S.; BARZEGAR-JALALI, M. Predicting human intestinal permeability using single-pass intestinal perfusion in rat. J. Pharm. Pharm. Sci., v.10, p.368-379, 2007.). A number of different in vivo, in situ and in vitro methods have emerged to determine the intestinal permeability of drugs and their mechanism of absorption. These include diffusion studies with intestinal segments from various species (e.g. rat and rabbit) or with cultured cell monolayers (e.g. Caco-2 cells); uptake studies in brush-border membrane vesicles prepared from intestinal segments of various species; and the single-pass intestinal perfusion (SPIP) in small mammals, most commonly rats (Artursson, Karlsson, 1991ARTURSSON, P.; KARLSSON, J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun., v.175, p.880-885, 1991.; Hillgren et al., 1995HILLGREN, K.M.; KATO, A.; BORCHARDT, R.T. In vitro systems for studying intestinal drug absorption. Med. Res. Rev., v.15, p.83-109, 1995.; Salphati et al., 2001SALPHATI, L.; CHILDERS, K.; PAN, L.; TSUTSUI, K.; TAKAHASHI, L. Evaluation of a single-pass intestinal-perfusion method in rat for the prediction of absorption in man. J. Pharm. Pharmacol., v.53, p.1007-1013, 2001.). Among these, single-pass intestinal perfusion (SPIP) provides conditions closer to what is faced following oral administration (Cook, Shenoy, 2003COOK, T.J.; SHENOY, S.S. Intestinal permeability of chlorpyrifos using the single-pass intestinal perfusion method in the rat. Toxicology, v.184, p.125-133, 2003.; Jeong et al., 2004JEONG, E.J.; LIU, Y.; LIN, H.; HU, M. In situ single-pass perfused rat intestinal model for absorption and metabolism. In: YAN, Z.; CALDWELL, G.W. (Eds.). Optimization in drug discovery: in vitro methods. New York: Humana Press, 2004. p.65-76.). This technique has low sensitivity to pH variations due to the preserved microclimate above the epithelial cells, and it maintains an intact blood supply to the intestine as well as providing the unique advantages of experimental control (e.g. compound concentration and intestinal perfusion rate), and the ability to study regional differences (Song et al., 2006SONG, N.N.; LI, Q.S.; LIU, C.X. Intestinal permeability of metformin using single-pass intestinal perfusion in rats. World J. Gastroenterol., v.12, p.4064-4070, 2006.; Wu et al., 2004WU, Y.; LOPER, A.; LANDIS, E.; HETTRICK, L.; NOVAK, L.; LYNN, K.; CHEN, C.; THOMPSON, K.; HIGGINS, R.; BATRA, U.; SHELUKAR, S.; KWEI, G.; STOREY, D. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: a Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int. J. Pharm., v.285, p.135-146, 2004.; Zakeri-Milani et al., 2009bZAKERI-MILANI, P.; VALIZADEH, H.; TAJERZADEH, H.; ISLAMBULCHILAR, Z. The utility of rat jejunal permeability for biopharmaceutics classification system. Drug Dev. Ind. Pharm., v.35, p.1496-1502, 2009b.). Clarithromycin (CLA) 6-O-methylerythromycin, is a semi-synthetic macrolide antibiotic with a broad antibacterial spectrum which is used in many infectious conditions like upper and lower respiratory tracts infections, skin, ear and other soft tissues infections, caused by different bacterial groups (Alkhalidi et al., 2008ALKHALIDI, B.A.; TAMIMI, J.J.; SALEM, I.I; IBRAHIM, H.; SALLAM, A.A. Assessment of the bioequivalence of two formulations of clarithromycin extended-release 500-mg tablets under fasting and fed conditions: a single-dose, randomized, open-label, two-period, two-way crossover study in healthy Jordanian male volunteers. Clin. Ther., v.30, p.1831-1843, 2008.; Gomez-Burgaz et al., 2009GOMEZ-BURGAZ, M.; TORRADO, G.; TORRADO, S. Characterization and superficial transformations on mini-matrices made of interpolymer complexes of chitosan and carboxymethylcellulose during in vitro clarithromycin release. Eur. J. Pharm. Biopharm., v.73, p.130-139, 2009.; Lu et al., 2008GARVER, E.; HUGGER, E.D.; SHEARN, S.P.; RAO, A.; DAWSON, P.A.; DAVIS, C.B.; HAN, C. Involvement of intestinal uptake transporters in the absorption of azithromycin and clarithromycin in the rat. Drug Metab. Dispos., v.36, p.2492-2498, 2008.). Clarithromycin is also the drug of choice to treat peptic ulcer as H. pylori resistance rate is much lower for clarithromycin as compared to other antibiotics like amoxicillin and tetracycline (Jain et al., 2006JAIN, N.; UMA MAHESHWARI, R.; RAMTEKE, S. Clarithromycin based oral sustained release nanoparticulate drug delivery system. Indian J. Pharm. Sci., v.68, p.479-484, 2006.; Zakeri-Milani et al., 2005ZAKERI-MILANI, P.; BARZEGAR-JALALI, M.; TAJERZADEH, H.; AZARMI, Y.; VALIZADEH, H. Simultaneous determination of naproxen, ketoprofen and phenol red in samples from rat intestinal permeability studies: HPLC method development and validation. J. Pharm. Biomed. Anal., v.39, p.624-630, 2005.). However, like many other macrolide antibiotics, clarithromycin exhibits poor absorption and low bioavailability when administered orally (Inoue et al., 2007INOUE, Y.; YOSHIMURA, S.; TOZUKA, Y.; MORIBE, K.; KUMAMOTO, T.; ISHIKAWA, T.; YAMAMOTO, K. Application of ascorbic acid 2-glucoside as a solubilizing agent for clarithromycin: solubilization and nanoparticle formation. Int. J. Pharm., v.331, p.38-45, 2007.). It is extensively metabolized by CYP3A (which is highly expressed in the gastrointestinal tract) to 14-hydroxyclarithromycin and N-desmethylclarithromycin and exhibits nonlinear pharmacokinetics, demonstrated by reduced clearance with increasing doses. Although these metabolites may also inactivate CYP3A, they occur at low systemic concentrations in vivo such that circulating metabolites will probably have an insignificant effect on CYP3A (Quinney et al., 2010QUINNEY, S.K.; ZHANG, X.; LUCKSIRI, A.; GORSKI, J.C.; LI, L.; HALL, S.D. Physiologically based pharmacokinetic model of mechanism-based inhibition of CYP3A by clarithromycin. Drug Metab. Dispos. v.38, p.241-248, 2010.). Therefore it is desirable to design CLA-loaded biodegradable nanoparticles with improved physicochemical properties and antibacterial activity against intracellular bacteria as well as an enhanced intestinal permeability and thereafter an increased bioavailability. The objective of the present study was to determine the permeability of clarithromycin previously (Mohammadi et al., 2011MOHAMMADI, G.; NOKHODCHI, A.; BARZEGAR-JALALI, M.; LOTFIPOUR, F.; ADIBKIA, K.; EHYAEI, N.; VALIZADEH, H. Physicochemical and anti-bacterial performance characterization of clarithromycin nanoparticles as colloidal drug delivery system. Colloids Surf.B Biointerfaces, v.88, p.39-44, 2011.) prepared as PLGA nanoparticles in comparison with pure drug solution using single-pass intestinal perfusion technique in rats and to investigate whether a nanoparticulate formulation could improve intestinal permeability of the compound.
MATHERIAL AND METHODS
Material
Clarithromycin powder was obtained from Dr Reddy’s Pharmaceutical Company, India. Poly (d,l-lactide-co-glycolide) (PLGA) (50:50 d,l-lactide:glycolide) with average molecular weight of 12,000 g/mol (Resomer RG 502), was purchased from Boehringer Ingelheim, Germany. Micrococcus luteus ATCC 9341 was purchased from Pasteur Institute, Iran. Poly vinyl alcohol, PVA, with molecular weight of 95000 (Acros Organics, Geel, Belgium) were used. Phenol red was purchased from Sigma Chemical Co. (St. Louis, MO). KH2PO4, NaH2PO4, Na2HPO4, Orthophosphoric acid, NaOH, and NaCI were purchased from Merck (Darmstadt, Germany). All other materials were of analytical or HPLC grade and obtained from Merck (Darmstadt, Germany).
Preparation of solutions
The perfusion buffer consisted of 5.77 g/L Na2HPO4 (anhydrous), 4.085 g/L NaH2PO4.2H2O, and 7 g/L NaCl. The pH of prepared buffer was adjusted to 7.2. Phenol red (0.7 mM) and metoprolol (0.07 mM) were added to the solution in all experiments as non-absorbable marker and internal standard respectively. Phenol red (0.7 mM) was added to the solution as a non-absorbable marker in all experiments. Control drug solutions were prepared in perfusion buffer (PBS) to obtain clarithromycin concentrations of 150, 250 and 400 µg/mL.
Preparation of nanoparticles
Nanoparticles were prepared by nano-precipitation according to the modified quasi emulsion solvent diffusion technique. CLA and PLGA powders (with 1:3 ratio) were co-dissolved in internal phase containing acetone, 2.5 mL, at room temperature (25 °C). The resulting organic solution was injected at the constant rate of 0.5 mL/min in aqueous phase (40 mL) containing PVA 95000 (2% w/v), as a stabilizing agent (Salem, Duzgunes, 2003). The process was carried out under homogenization for 5 min using the Silent Crusher M (Heidolph, Germany). The agitation speed was 13000 rpm in an ice-water bath. Organic phase was eliminated at room temperature under stirring for 12 hours. The final nano-suspension was centrifuged (Beckman Centrifuge, AvantiTMJ-25, USA) at 14000 rpm for 30 min and the precipitated nanoparticles were washed twice with water, using the previously described centrifugation approach, and then lyophilized using a lyophilizer (Christ Alpha 1-4; Germany). Final dry powder was taken out for further investigations. The nanoparticle suspensions were prepared to obtain concentrations of 150, 250 and 400 µg/mL (Mohammadi et al., 2011MOHAMMADI, G.; NOKHODCHI, A.; BARZEGAR-JALALI, M.; LOTFIPOUR, F.; ADIBKIA, K.; EHYAEI, N.; VALIZADEH, H. Physicochemical and anti-bacterial performance characterization of clarithromycin nanoparticles as colloidal drug delivery system. Colloids Surf.B Biointerfaces, v.88, p.39-44, 2011.).
Physicochemical evaluation of nanoparticles
Evaluation of the physicochemical properties of the prepared nanoparticles was performed using encapsulation efficiency and dissolution studies, particle size analysis, zeta potential determination, differential scanning calorimetry and Fourier-transform infrared spectroscopy. The nanoparticle production yield was determined from the mass ratio of the prepared nanoparticle to the initially added PLGA and drug (Zakeri-Milani et al., 2013ZAKERI-MILANI, P.; LOVEYMI, B.D.; JELVEHGARI, M.; VALIZADEH, H. The characteristics and improved intestinal permeability of vancomycin PLGA-nanoparticles as colloidal drug delivery system. Colloids Surf. B Biointerfaces, v.103, p.174-181, 2013.). The experiment was carried out using 3 samples equivalent to 5 mg drug. The mean particle-size values were measured using a laser diffraction particle-size analyzer (Sald 2101, Shimadzu, Japan) equipped with Wing software (version 1.20). Scanning electron microscopy (SEM) (LEO 440i, Leo Electron Microscopy Ltd., Cambridge, UK) at an accelerating voltage of 20 kV was used to examine the morphology of the nanoparticles. The particle surface charge was quantified as zeta potential using a Zetasizer 4 (Malvern Instr., UK). Thermograms of CLA, PLGA, nanoparticles and corresponding physical mixture of the drug with the polymer were recorded on a DSC-60 (Shimadzu, Kyoto, Japan). For drug release study, the prepared nanoparticles were placed in the vessels containing 250 mL phosphate buffer (pH 6). The vessels were incubated at 37oC with continuous orbital mixing (50 rpm). At specified time intervals, 3 mL of medium was removed using a glass syringe fitted to cellulose acetate membrane, 25 mm diameter and 20 nm pore diameter (Whatman, UK). After each sampling, an equal volume of fresh dissolution media was passed through the same filtration assembly to replace the withdrawn aliquots. The cumulative amount of the released CLA was calculated considering the replaced volume of the dissolution medium and the cumulative percentage of the released drug was plotted versus time. The mean calculated values were obtained from 3 replicates (Mohammadi et al., 2011MOHAMMADI, G.; NOKHODCHI, A.; BARZEGAR-JALALI, M.; LOTFIPOUR, F.; ADIBKIA, K.; EHYAEI, N.; VALIZADEH, H. Physicochemical and anti-bacterial performance characterization of clarithromycin nanoparticles as colloidal drug delivery system. Colloids Surf.B Biointerfaces, v.88, p.39-44, 2011.).
Single-pass intestinal perfusion experiment
Nanoparticles were prepared by nano-precipitation according to the modified quasi emulsion solvent diffusion technique. CLA and PLGA powders (with 1:3 ratio) were co-dissolved in internal phase containing acetone, 2.5 mL, at room temperature (25 °C). The resulting organic solution was injected at the constant rate of 0.5 mL/min in aqueous phase (40 mL) containing PVA 95000 (2% w/v), as a stabilizing agent (Salem, Duzgunes, 2003). The process was carried out under homogenization for 5 min using the Silent Crusher M (Heidolph, Germany). The agitation speed was 13000 rpm in an ice-water bath. Organic phase was eliminated at room temperature under stirring for 12 hours. The final nano-suspension was centrifuged (Beckman Centrifuge, AvantiTMJ-25, USA) at 14000 rpm for 30 min and the precipitated nanoparticles were washed twice with water, using the previously described centrifugation approach, and then lyophilized using a lyophilizer (Christ Alpha 1-4; Germany). Final dry powder was taken out for further investigations. The nanoparticle suspensions were prepared to obtain concentrations of 150, 250 and 400 µg/mL (Mohammadi et al., 2011MOHAMMADI, G.; NOKHODCHI, A.; BARZEGAR-JALALI, M.; LOTFIPOUR, F.; ADIBKIA, K.; EHYAEI, N.; VALIZADEH, H. Physicochemical and anti-bacterial performance characterization of clarithromycin nanoparticles as colloidal drug delivery system. Colloids Surf.B Biointerfaces, v.88, p.39-44, 2011.).
Microbial analysis of clarithromycin in the samples
Microorganism and inoculums standardization
Micrococcus luteus ATCC 9341 was purchased in lyophilized form (Pasteur Institute, Iran) and activated in trypticase soy broth medium. Fifty microliters of the growth medium was transferred into antibiotic agar medium I (24 h before assay) and incubated at 35 °C for one day. The bacterial growth culture was diluted with a 0.9% w/v saline solution, in order to reach 30% turbidity at 580 nm, and the resultant bacterial suspension was then used as culturing inoculums.
Well diffusion assay
Microbiological assay of clarithromycin in the samples was performed using an agar well diffusion procedure (USP, 2008). Briefly, the assay plate contained 25 mL of antibiotic agar I inoculated with the bacterial inoculums. Wells of 6mm diameter were punched and filled with 100 µL of calibration samples or test samples. After 24 h of incubation at 35 °C, the diameter of the inhibition zone was measured. The method was validated by determination of the following operational characteristics: linearity, precision and accuracy (Mohammadi et al., 2011MOHAMMADI, G.; NOKHODCHI, A.; BARZEGAR-JALALI, M.; LOTFIPOUR, F.; ADIBKIA, K.; EHYAEI, N.; VALIZADEH, H. Physicochemical and anti-bacterial performance characterization of clarithromycin nanoparticles as colloidal drug delivery system. Colloids Surf.B Biointerfaces, v.88, p.39-44, 2011.). The linearity was evaluated using the linear regression analysis which was calculated by the least squares regression method. Clarithromycin calibration standards were prepared at concentrations of 500, 450, 350, 250, 150 and 50 µg/mL. Each level was made in triplicate and employed on the well diffusion assay method described above. The precision of the assay method was determined by evaluating repeatability (intra-assay) and intermediate precision (inter-assay), and expressed as the relative standard deviation (RSD) of four quality control samples. The accuracy was determined by adding known amounts of clarithromycin reference substance (quality control samples) at the beginning of the process, followed by calculation of the value: measured value/nominal value × 100.
Liquid chromatographic conditions for analysis of phenol red
For analysis of phenol red in the samples, Shimadzu HPLC system (Shimadzu, Kyoto, Japan) comprising an LC-10ADvp pump and a variable wavelength ultraviolet spectrophotometric detector (SPD-10Avp) set at 430 nm was used.
The phenol red concentrations in the perfusion buffer were determined using a previously developed HPLC method in which the mobile phase consisting of a mixture of 55% methanol and 45% of 0.05 mol/L KH2P04 aqueous solution (adjusted to pH 2.6) was pumped into a Shimpack VP-ODS 5 µm 4.6 × 250 mm with a Shimpack VP-ODS 5 µm 4.6 ×50 mm guard column at a flow rate of 1 mL/min. Under these conditions the phenol red retention time was found to be 3 min (Swenson et al., 1994SWENSON, E.S.; MILISEN, W.B.; CURATOLO, W. Intestinal permeability enhancement: efficacy, acute local toxicity, and reversibility. Pharm. Res., v.11, p.1132-1142, 1994.; Valizadeh et al., 2006VALIZADEH, H.; ZAKERI-MILANI, P.; ISLAMBULCHILAR, Z.; TAJERZADEH, H. A simple and rapid high-performance liquid chromatography method for determining furosemide, hydrochlorothiazide, and phenol red: applicability to intestinal permeability studies. J. AOAC Int., v.89, p.88-93, 2006.; Zakeri-Milani et al., 2005ZAKERI-MILANI, P.; BARZEGAR-JALALI, M.; TAJERZADEH, H.; AZARMI, Y.; VALIZADEH, H. Simultaneous determination of naproxen, ketoprofen and phenol red in samples from rat intestinal permeability studies: HPLC method development and validation. J. Pharm. Biomed. Anal., v.39, p.624-630, 2005.). Class VP software was used for data acquisition and processing.
Data Analysis
Effective permeability coefficients (Peff) were calculated after correcting the steady-state outlet concentrations for water flux based on the ratio of inlet and outlet concentrations of an un-absorbable marker, phenol red. The SPIP technique performed in the present study for estimating intestinal permeability coefficient uses a mass balance approach. Two models namely Well-stirred model (i.e. mixing tank model) and Parallel tube model (i.e. complete radial mixing model) are used to explain this approach. Steady state was reached about 40 min after the beginning of the perfusion and was confirmed by plotting the ratio of the outlet to inlet concentrations versus time. The representative results for CLA 250 μg/mL in perfusion solution are plotted in Figure 1. Assuming the parallel tube model, the Peff values were calculated using the following equation:
Peff= -Qin [Cout/Cin]/2πrl
where Q is the perfusion buffer flow rate (0.2 mL/ min), Cout/Cin is the ratio of the outlet concentration and inlet concentration of clarithromycin that has been adjusted for water transport during the perfusion, r is the radius of the intestinal segment (0.18 cm) and l is the length of the intestinal segment. Values were indicated as mean ± SD for permeability in four independent rats. Statistical difference between the permeabilities of different concentrations of clarithromycin in the form of solution and nanoparticle suspension was evaluated by two-tailed Student’s t-test. P-values<0.05 were considered significant.
RESULTS AND DISCUSSION
Micrococcus luteus was selected for performing the antibacterial assay as it is a non-pathogenic bacterium and has good sensitivity to clarithromycin (Mohammadi et al., 2011MOHAMMADI, G.; NOKHODCHI, A.; BARZEGAR-JALALI, M.; LOTFIPOUR, F.; ADIBKIA, K.; EHYAEI, N.; VALIZADEH, H. Physicochemical and anti-bacterial performance characterization of clarithromycin nanoparticles as colloidal drug delivery system. Colloids Surf.B Biointerfaces, v.88, p.39-44, 2011.). The six point linearity curves for clarithromycin were constructed in the range of 50-500 µg/mL. These concentration ranges were selected based on the drug concentrations used in the permeability studies. The regression analysis of the obtained responses generated a linear curve with a correlation coefficient of 0.992. Nanoparticles with efficient loading of clarithromycin and submicron size range were obtained. Loading efficiencies of the obtained particles were calculated using the equation shown below:
Loading efficiency (%) = (actual drug content in nanoparticles/theoretical drug content) × 100.
Loading efficiencies for three different concentrations of drug (400, 250 and 150 µg/mL) were between 71.78% - 98.8% (Table I). Poor entrapment of drug in the applied concentration of 400 µg/mL could be due to the low aqueous solubility of both polymer and CLA (practically insoluble) in water. Higher loading efficiency was obtained with decreasing the initially applied amount of the drug.
According to the results of particle size analysis and SEM studies (Figure 1), the obtained nanoparticles were between 200 to 500 nm in size with narrow size distribution, and spherical shape. The mean particle size was found to be 305 ± 134 nm.
Zeta potential of the nanoparticles, intact polymer and CLA were -20.32 ± 2.84 and 1.47 ± 1.61, and -14.26 ± 1.92 mV, respectively. Intact CLA powder had an endothermic peak corresponding to its melting point at 231.34 oC, there was no distinct CLA melting endotherm in the nanoparticle, suggesting a complete amorphization of the drug in the prepared nanoparticles (Figure 2).
DSC curves of the intact clarithromycin (CLA), PLGA, their physical mixture (PM) and clarithromycin loaded nanoparticles (NANO) of 1:3 drug to polymer ratio.
The time required for 50% of drug dissolved (t50%) which is inversely related to the dissolution rate are 30 min for CLA and 180 min for nanoparticles. The presence of insoluble polymer in the nanoparticles matrix body reduces the water penetration, hence dissolution and diffusion. A very slow release pattern for nanoparticles was seen after an initial burst within 4 h. 59% of drug was released during the first 4 hours from nanoparticles. This may be attributed to the dissolution of the drug that is poorly entrapped in the polymer matrix, while the slower and continuous release may be ascribed to the diffusion of the drug localized in the PLGA core of the nanoparticles. In SPIP experiments steady state was reached about 40 min after the beginning of the perfusion and was confirmed by plotting the ratio of the outlet to inlet concentrations versus time. The representative results for CLA 250 μg/mL in perfusion solution are plotted in Figure 3.
Plot of concentration ratio of the inlet and outlet tubing (Cin/Cout) vs. time for CLA (250 μg/mL) perfusion study (n=4, error bars represent SD).
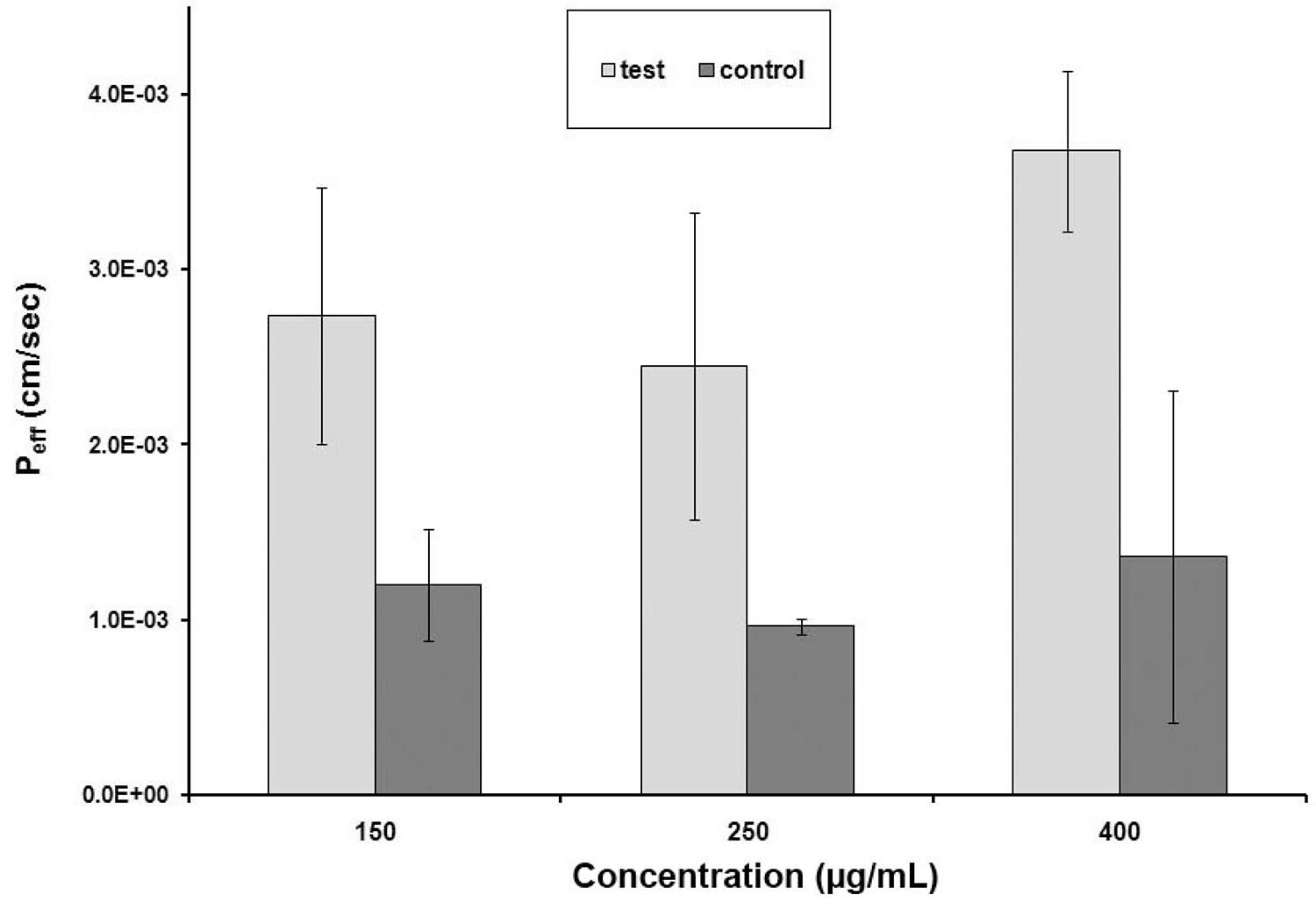
The intestinal permeability of clarithromycin in three different concentrations was determined in rat jejunum using the in situ single-pass perfusion technique. The samples were analyzed by the above mentioned method. Prior to the in situ study, stability assessment was performed to evaluate the stability of the test drug in blank perfusion samples by simulating the same conditions which occurred during the study. The results showed that drug was sufficiently stable throughout the duration of experiment (120 min) in all investigated solutions. The mean effective permeability coefficients of clarithromycin solution in concentrations of 150, 250 and 400 µg/mL in perfusion solution were found to be 1.20(±0.32)×10-3 cm/s, 9.62(±0.46)×10-4 cm/s, and 1.36(±0.946)×10-3 cm/s respectively. The corresponding values at the same concentration of nanoparticles were found to be 2.74(±073)×10-3 cm/s, 2.45(±0. 877)×10-3 cm/s, and 3.68(±0.46)×10-3 cm/s respectively. The effective permeability of clarithromycin from nanoparticle suspension was 2.28 fold enhanced in comparison to the solution form at the concentration of 150 µg/mL whereas these enhancement ratios were 2.54 and 2.70 in the case of 250 and 400 µg/mL samples, respectively. The obtained data suggests a slight increase in the enhancement ratio by increasing the concentration of drug within nanoparticles. As it is shown in Figure 4, the two-tailed Student’s t-test showed that the intestinal permeability of clarithromycin was significantly increased in the nanoparticle suspension form in all four clarithromycin concentrations used (P < 0.05).
Intestinal permeability of clarithromycin in control and test samples at three concentration levels. Values are the mean ± SD of four determinations.
This finding is in agreement with the previously published literature about the intestinal uptake of nano sized particles. For instance in one study the results of in vitro permeation experiments by Ussing chambers has shown that the permeability of tacrolimus was enhanced significantly in the form of nanoparticles in comparison with its solution in both healthy and inflamed tissues (Lamprecht et al., 2005LAMPRECHT, A.; YAMAMOTO, H.; TAKEUCHI, H.; KAWASHIMA, Y. Nanoparticles enhance therapeutic efficiency by selectively increased local drug dose in experimental colitis in rats. J. Pharmacol. Exp. Ther., v.315, p.196-202, 2005.). In another study, PLGA nanoparticles loaded with curcumin was designed and prepared and the comparison of in situ intestinal permeability and also the bioavailability of these nanoparticles with that of native curcumin showed that encapsulating the drug in PLGA polymers could enhance its intestinal permeability as well as its in vivo bioavailability (Xie et al., 2011XIE, X.; TAO, Q.; ZOU, Y.; ZHANG, F.; GUO, M.; WANG, Y.; WANG, H.; ZHOU, Q.; YU, S. PLGA nanoparticles improve the oral bioavailability of curcumin in rats: characterizations and mechanisms. J. Agric. Food Chem., v.59, p.9280-9289, 2011.). In a similar study, the cyclosporine nanoparticles showed significantly higher intestinal uptake of around 90% as compared to Sandimmune Neoral®, which showed around 55% and Cyclosporine sodium CMC suspension with an uptake of only 10% (Italia et al., 2007ITALIA, J.L.; BHATT, D.K.; BHARDWAJ, V.; TIKOO, K.; KUMAR, M.N. PLGA nanoparticles for oral delivery of cyclosporine: nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral. J. Control. Release, v.119, p.197-206, 2007.).
It has been demonstrated that the effect of delivering the drug in the nanoparticle form on bioavailability are primarily based on the fundamentals that nanonization increases the contact surface area of drugs. Drug nanonization would result in an increase in adhesion surface area between intestinal epithelial cells and nanoparticles as well as an increased dissolution rate and also an increase in saturation solubility that favors increased concentration gradient between intestinal epithelial cells and the mesenteric circulation beneath (Mohanraj, Chen, 2006MOHANRAJ, V.; CHEN, Y. Nanoparticles - a review. Trop. J. Pharm. Res., v.5, p.561-573, 2006.). The results of such studies showed that PLGA may have bioadhesive properties and bind with the mucosa of the gastrointestinal tract. This may increase the residency time and enhance drug absorption due to intimate contact with epithelium cells. It was also reported that PLGA as a drug carrier moderates the P-gp effect and MDR reversal activity, however, the P-gp inhibition mechanism of PLGA, which mainly involves changing the fluidity of the cellular membrane, inhibiting P-gp ATPase, and reducing P-gp expression, remains unclear (Xie et al., 2011XIE, X.; TAO, Q.; ZOU, Y.; ZHANG, F.; GUO, M.; WANG, Y.; WANG, H.; ZHOU, Q.; YU, S. PLGA nanoparticles improve the oral bioavailability of curcumin in rats: characterizations and mechanisms. J. Agric. Food Chem., v.59, p.9280-9289, 2011.). In general, the gastrointestinal absorption of macromolecules and particulate materials involves either paracellular route or endocytotic pathway. Using polymers such as chitosan, starch or poly (acrylate) can increase the paracellular permeability of macromolecules. Endocytotic pathway for absorption of nanoparticles is either by receptor-mediated endocytosis, that is, active targeting, or adsorptive endocytosis which does not need any ligands. This process is initiated by an unspecific physical adsorption of material to the cell surface by electrostatic forces such as hydrogen bonding or hydrophobic interactions. Adsorptive endocytosis depends primarily on the size and surface properties of the material. This shows that a combination of size, surface charge and hydrophilicity play a major role in affinity (Damge et al., 1996DAMGE, C.; APRAHAMIAN, M.; MARCHAIS, H.; BENOIT, J.P.; PINGET, M. Intestinal absorption of PLAGA microspheres in the rat. J. Anat., v.189 (Pt 3), p.491-501, 1996.; Kotze et al., 1998KOTZE, A.F.; LUESSEN, H.L.; DE LEEUW, B.J.; DE BOER, A.G.; VERHOEF, J.C.; JUNGINGER, H.E. Comparison of the effect of different chitosan salts and N-trimethyl chitosan chloride on the permeability of intestinal epithelial cells (Caco-2). J. Control. Release, v.51, p.35-46, 1998.; Win, Feng, 2005WIN, K.Y.; FENG, S.S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials, v.26, p.2713-2722, 2005.).
Although clarithromycin exhibits undesirable physicochemical and biopharmaceutical properties (e.g., large molecular weight, extensive potential for hydrogen bonding, high polar surface area, substrates for efflux transporter P-glycoprotein), based on the obtained results it showed high intestinal permeability in rats. The drug was previously reported to have moderate to excellent oral bioavailability in preclinical species and humans (Lan et al., 2009LAN, T.; RAO, A.; HAYWOOD, J.; DAVIS, C.B.; HAN, C.; GARVER, E.; DAWSON, P.A. Interaction of macrolide antibiotics with intestinally expressed human and rat organic anion-transporting polypeptides. Drug Metab. Dispos., v.37, p.2375-2382, 2009.). A potential explanation for this paradox is that intestinal transporters may facilitate CLA absorption. Previous studies in rats suggested that oral absorption of azithromycin and clarithromycin is mediated by an Oatp and/or other rifamycin SV-sensitive intestinal transporter (Garver et al., 2008GARVER, E.; HUGGER, E.D.; SHEARN, S.P.; RAO, A.; DAWSON, P.A.; DAVIS, C.B.; HAN, C. Involvement of intestinal uptake transporters in the absorption of azithromycin and clarithromycin in the rat. Drug Metab. Dispos., v.36, p.2492-2498, 2008.). This indicates that the drug may be involved in active transport mechanisms which causes the drug fell outside the lipiniski ‘rule of 5’ (Doppenschmitt et al., 1999DOPPENSCHMITT, S.; SPAHN-LANGGUTH, H.; REGARDH, C.G.; LANGGUTH, P. Role of P-glycoprotein-mediated secretion in absorptive drug permeability: an approach using passive membrane permeability and affinity to P-glycoprotein. J. Pharm. Sci., v.88, p.1067-1072, 1999.).
CONCLUSION
The results of the present study suggest that formulating clarithromycin as nanoparticles, could improve its intestinal permeability in comparison with the pure drug solution. This could be at least partly, due to the contribution of PLGA in alteration of the intestinal permeability.
ACKNOWLEDGMENT
The authors would like to thank the authorities of the Research Center for Pharmaceutical Nanotechnology, Tabriz University of Medical Sciences For their support. This article is based on a thesis submitted for PharmD degree (No. 3492) in Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran.
REFERENCES
- ALKHALIDI, B.A.; TAMIMI, J.J.; SALEM, I.I; IBRAHIM, H.; SALLAM, A.A. Assessment of the bioequivalence of two formulations of clarithromycin extended-release 500-mg tablets under fasting and fed conditions: a single-dose, randomized, open-label, two-period, two-way crossover study in healthy Jordanian male volunteers. Clin. Ther., v.30, p.1831-1843, 2008.
- ARAYNE, M.S.; SULTANA, N. Review: nanoparticles in drug delivery for the treatment of cancer. Pak. J. Pharm. Sci., v.19, p.258-268, 2006.
- ARTURSSON, P.; KARLSSON, J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun., v.175, p.880-885, 1991.
- CAI, Z.; WANG, Y.; ZHU, L.J.; LIU, Z.Q. Nanocarriers: a general strategy for enhancement of oral bioavailability of poorly absorbed or pre-systemically metabolized drugs. Curr. Drug Metab., v.11, p.197-207, 2010.
- COOK, T.J.; SHENOY, S.S. Intestinal permeability of chlorpyrifos using the single-pass intestinal perfusion method in the rat. Toxicology, v.184, p.125-133, 2003.
- DAMGE, C.; APRAHAMIAN, M.; MARCHAIS, H.; BENOIT, J.P.; PINGET, M. Intestinal absorption of PLAGA microspheres in the rat. J. Anat., v.189 (Pt 3), p.491-501, 1996.
- DOPPENSCHMITT, S.; SPAHN-LANGGUTH, H.; REGARDH, C.G.; LANGGUTH, P. Role of P-glycoprotein-mediated secretion in absorptive drug permeability: an approach using passive membrane permeability and affinity to P-glycoprotein. J. Pharm. Sci., v.88, p.1067-1072, 1999.
- FLORENCE, A.T. Issues in oral nanoparticle drug carrier uptake and targeting. J. Drug Target., v.12, p.65-70, 2004.
- GARVER, E.; HUGGER, E.D.; SHEARN, S.P.; RAO, A.; DAWSON, P.A.; DAVIS, C.B.; HAN, C. Involvement of intestinal uptake transporters in the absorption of azithromycin and clarithromycin in the rat. Drug Metab. Dispos., v.36, p.2492-2498, 2008.
- GOMEZ-BURGAZ, M.; TORRADO, G.; TORRADO, S. Characterization and superficial transformations on mini-matrices made of interpolymer complexes of chitosan and carboxymethylcellulose during in vitro clarithromycin release. Eur. J. Pharm. Biopharm., v.73, p.130-139, 2009.
- HILLGREN, K.M.; KATO, A.; BORCHARDT, R.T. In vitro systems for studying intestinal drug absorption. Med. Res. Rev., v.15, p.83-109, 1995.
- INOUE, Y.; YOSHIMURA, S.; TOZUKA, Y.; MORIBE, K.; KUMAMOTO, T.; ISHIKAWA, T.; YAMAMOTO, K. Application of ascorbic acid 2-glucoside as a solubilizing agent for clarithromycin: solubilization and nanoparticle formation. Int. J. Pharm., v.331, p.38-45, 2007.
- ITALIA, J.L.; BHATT, D.K.; BHARDWAJ, V.; TIKOO, K.; KUMAR, M.N. PLGA nanoparticles for oral delivery of cyclosporine: nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral. J. Control. Release, v.119, p.197-206, 2007.
- JAIN, N.; UMA MAHESHWARI, R.; RAMTEKE, S. Clarithromycin based oral sustained release nanoparticulate drug delivery system. Indian J. Pharm. Sci., v.68, p.479-484, 2006.
- JEONG, E.J.; LIU, Y.; LIN, H.; HU, M. In situ single-pass perfused rat intestinal model for absorption and metabolism. In: YAN, Z.; CALDWELL, G.W. (Eds.). Optimization in drug discovery: in vitro methods. New York: Humana Press, 2004. p.65-76.
- KOTZE, A.F.; LUESSEN, H.L.; DE LEEUW, B.J.; DE BOER, A.G.; VERHOEF, J.C.; JUNGINGER, H.E. Comparison of the effect of different chitosan salts and N-trimethyl chitosan chloride on the permeability of intestinal epithelial cells (Caco-2). J. Control. Release, v.51, p.35-46, 1998.
- LAMPRECHT, A.; YAMAMOTO, H.; TAKEUCHI, H.; KAWASHIMA, Y. Nanoparticles enhance therapeutic efficiency by selectively increased local drug dose in experimental colitis in rats. J. Pharmacol. Exp. Ther., v.315, p.196-202, 2005.
- LAN, T.; RAO, A.; HAYWOOD, J.; DAVIS, C.B.; HAN, C.; GARVER, E.; DAWSON, P.A. Interaction of macrolide antibiotics with intestinally expressed human and rat organic anion-transporting polypeptides. Drug Metab. Dispos., v.37, p.2375-2382, 2009.
- LU, Y.; WANG, Y.; TANG, X. Formulation and thermal sterile stability of a less painful intravenous clarithromycin emulsion containing vitamin E. Int. J. Pharm., v.346, p.47-56, 2008.
- MOHAMMADI, G.; NOKHODCHI, A.; BARZEGAR-JALALI, M.; LOTFIPOUR, F.; ADIBKIA, K.; EHYAEI, N.; VALIZADEH, H. Physicochemical and anti-bacterial performance characterization of clarithromycin nanoparticles as colloidal drug delivery system. Colloids Surf.B Biointerfaces, v.88, p.39-44, 2011.
- MOHANRAJ, V.; CHEN, Y. Nanoparticles - a review. Trop. J. Pharm. Res., v.5, p.561-573, 2006.
- MOINARD-CHECOT, D.; CHEVALIER, Y.; BRIANCON, S.; FESSI, H.; GUINEBRETIERE, S. Nanoparticles for drug delivery: review of the formulation and process difficulties illustrated by the emulsion-diffusion process. J. Nanosci. Nanotechnol., v.6, p.2664-2681, 2006.
- QUINNEY, S.K.; ZHANG, X.; LUCKSIRI, A.; GORSKI, J.C.; LI, L.; HALL, S.D. Physiologically based pharmacokinetic model of mechanism-based inhibition of CYP3A by clarithromycin. Drug Metab. Dispos. v.38, p.241-248, 2010.
- RAO, S.V.; YAJURVEDI, K.; SHAO, J. Self-nanoemulsifying drug delivery system (SNEDDS) for oral delivery of protein drugs: III. In vivo oral absorption study. Int. J. Pharm., v.362, p.16-19, 2008.
- SAHA, R.N.; VASANTHAKUMAR, S.; BENDE, G.; SNEHALATHA, M. Nanoparticulate drug delivery systems for cancer chemotherapy. Mol. Membr. Biol., v.27, p.215-231, 2010.
- SALEM, I.I.; DUZGUNES, N. Efficacies of cyclodextrin-complexed and liposome-encapsulated clarithromycin against Mycobacterium avium complex infection in human macrophages. Int. J. Pharm., v.250, p.403-414, 2003.
- SALPHATI, L.; CHILDERS, K.; PAN, L.; TSUTSUI, K.; TAKAHASHI, L. Evaluation of a single-pass intestinal-perfusion method in rat for the prediction of absorption in man. J. Pharm. Pharmacol., v.53, p.1007-1013, 2001.
- SONG, N.N.; LI, Q.S.; LIU, C.X. Intestinal permeability of metformin using single-pass intestinal perfusion in rats. World J. Gastroenterol., v.12, p.4064-4070, 2006.
- SWENSON, E.S.; MILISEN, W.B.; CURATOLO, W. Intestinal permeability enhancement: efficacy, acute local toxicity, and reversibility. Pharm. Res., v.11, p.1132-1142, 1994.
- USP (2008). United States Pharmacopoeia and National Formulary (USP 31-NF26). Rockville, 2008.
- VALIZADEH, H.; ZAKERI-MILANI, P.; ISLAMBULCHILAR, Z.; TAJERZADEH, H. A simple and rapid high-performance liquid chromatography method for determining furosemide, hydrochlorothiazide, and phenol red: applicability to intestinal permeability studies. J. AOAC Int., v.89, p.88-93, 2006.
- WIN, K.Y.; FENG, S.S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials, v.26, p.2713-2722, 2005.
- WU, Y.; LOPER, A.; LANDIS, E.; HETTRICK, L.; NOVAK, L.; LYNN, K.; CHEN, C.; THOMPSON, K.; HIGGINS, R.; BATRA, U.; SHELUKAR, S.; KWEI, G.; STOREY, D. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: a Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int. J. Pharm., v.285, p.135-146, 2004.
- XIE, X.; TAO, Q.; ZOU, Y.; ZHANG, F.; GUO, M.; WANG, Y.; WANG, H.; ZHOU, Q.; YU, S. PLGA nanoparticles improve the oral bioavailability of curcumin in rats: characterizations and mechanisms. J. Agric. Food Chem., v.59, p.9280-9289, 2011.
- ZAKERI-MILANI, P.; BARZEGAR-JALALI, M.; AZIMI, M.; VALIZADEH, H. Biopharmaceutical classification of drugs using intrinsic dissolution rate (IDR) and rat intestinal permeability. Eur. J. Pharm. Biopharm., v.73, p.102-106, 2009a.
- ZAKERI-MILANI, P.; BARZEGAR-JALALI, M.; TAJERZADEH, H.; AZARMI, Y.; VALIZADEH, H. Simultaneous determination of naproxen, ketoprofen and phenol red in samples from rat intestinal permeability studies: HPLC method development and validation. J. Pharm. Biomed. Anal., v.39, p.624-630, 2005.
- ZAKERI-MILANI, P.; LOVEYMI, B.D.; JELVEHGARI, M.; VALIZADEH, H. The characteristics and improved intestinal permeability of vancomycin PLGA-nanoparticles as colloidal drug delivery system. Colloids Surf. B Biointerfaces, v.103, p.174-181, 2013.
- ZAKERI-MILANI, P.; VALIZADEH, H.; ISLAMBULCHILAR, Z.; DAMANI, S.; MEHTARI, M. Investigation of the intestinal permeability of ciclosporin using the in situ technique in rats and the relevance of P-glycoprotein. Arzneimittelforschung, v.58, p.188-192, 2008.
- ZAKERI-MILANI, P.; VALIZADEH, H.; TAJERZADEH, H.; AZARMI, Y.; ISLAMBOLCHILAR, Z.; BARZEGAR, S.; BARZEGAR-JALALI, M. Predicting human intestinal permeability using single-pass intestinal perfusion in rat. J. Pharm. Pharm. Sci., v.10, p.368-379, 2007.
- ZAKERI-MILANI, P.; VALIZADEH, H.; TAJERZADEH, H.; ISLAMBULCHILAR, Z. The utility of rat jejunal permeability for biopharmaceutics classification system. Drug Dev. Ind. Pharm., v.35, p.1496-1502, 2009b.
Publication Dates
-
Publication in this collection
Jan-Mar 2014
History
-
Received
09 Apr 2013 -
Accepted
16 Sept 2013