Circulating Cell-Free DNA in Hepatocellular Carcinoma: Current Insights and Outlook
 Charlotte K. Y. Ng
Charlotte K. Y. Ng Giovan Giuseppe Di Costanzo
Giovan Giuseppe Di Costanzo Luigi M. Terracciano
Luigi M. Terracciano Salvatore Piscuoglio
Salvatore Piscuoglio- 1Institute of Pathology, University Hospital Basel, Basel, Switzerland
- 2Department of Biomedicine, Hepatology Laboratory, University of Basel, Basel, Switzerland
- 3Department of Transplantation – Liver Unit, Cardarelli Hospital, Naples, Italy
Over the past decade, the advancements in massively parallel sequencing have provided a new paradigm in biomedical research to uncover the genetic basis of human diseases. Integration of ‘omics information has begun transforming clinical management of cancer patients in terms of diagnostics and treatment options, giving rise to the era of precision medicine. Currently, nucleic acids for molecular profiling for patients diagnosed with hepatocellular carcinoma (HCC) are typically obtained from resected tumor materials or transplanted neoplastic liver and occasionally from biopsies. Given the intrinsic risks associated with such invasive procedures, circulating cell-free DNA (cfDNA) has been proposed as an alternative source for tumor DNA. Circulating cfDNA is a type of cell-free nucleic acid that derives from apoptotic, necrotic, as well as living eukaryotic cells. Importantly, the detection of abnormal forms of circulating cfDNA that originate from cancer cells provides a new tool for cancer detection, disease monitoring, and molecular profiling. Currently, cfDNA is beginning to be adopted into clinical practice as a non-invasive tool to monitor disease by tracking the evolution of disease-specific genetic alterations in several major cancer types. Moreover, cfDNA is demonstrating potential clinical value as a surrogate to assess the molecular makeup of tumors and to overcome the sampling biases inherent to intra-tumor genetic heterogeneity, especially in the metastatic setting. With the improvements in ‘omics and molecular biology techniques, coupled with the increasing understanding in the molecular pathogenesis of cancer, it can be anticipated that the detection and analysis of cfDNA will become more specific and sensitive and thus enable cfDNA analysis to be used as a diagnostic aid in patients with early-stage disease and perhaps even in a screening setting. In this review, we provide an overview of the latest findings on the role and potential utility of cfDNA analysis in the diagnosis, management, and screening of HCC.
Review Criteria
Review of the literature was performed in English using the PubMed database. Search terms used included “hepatocellular carcinoma,” “liver cancer,” “circulating cell free DNA,” “circulating tumor DNA,” “plasma,” “serum,” and “liquid biopsy.”
Introduction
Over the past decade, the advancements in next-generation sequencing (NGS) and high-throughput omics profiling have provided a new paradigm in biomedical research to uncover the epi/genetic basis of human diseases. Integration of omics information has begun to transform clinical management of cancer patients in terms of diagnostics, prevention, and treatment options, giving rise to the era of precision medicine. Compared to cancer types such as those of the breasts and the lungs, precision medicine for hepatocellular carcinoma (HCC) patients is lagging behind, owing to a number of HCC-specific challenges in clinical practice and the lack of treatment options.
Hepatocellular carcinoma accounts for >90% of liver cancers and was responsible for an estimated 782,000 new cancer cases and nearly 746,000 deaths in 2012 (1). While HCC is the sixth most common cause of cancer worldwide, it is the third most common cause of cancer deaths and has one of the highest mortality-to-incidence ratios (2). HCC usually occurs in cirrhotic livers, and the epidemiology of HCC shows marked variations between geographical regions and racial groups (3). In sub-Saharan Africa and south-east Asia, hepatitis B virus (HBV) chronic infection is endemic and accounts for the majority of HCCs diagnosed (2). In Western populations, the rising incidence of HCC is in part driven by the increasing prevalence of chronic liver diseases associated with hepatitis C virus (HCV) infection, alcohol consumption, diabetes, obesity, and non-alcoholic fatty liver disease (2, 4, 5). Other known risk factors of HCC include hereditary diseases such as hemochromatosis and α-1-antitrypsin deficiency, exposure to aflatoxin B1 through dietary consumption, smoking, and the male gender (2). The 5-year cumulative risk for HCC in patients with cirrhosis ranges between 5 and 30%, depending on the cause (with the highest risk among those infected with HCV), region or ethnic group (17% in the United States and 30% in Japan), and stage of cirrhosis (with the highest risk among patients with decompensated disease) (6).
The stagnating prognosis of HCC is contributed in part by the limited treatment options. For early-stage HCC, curative treatment options involve resection and/or liver transplantation, with >50% 5-year overall survival. By contrast, for patients with late-stage unresectable disease or ineligible for transplantation, 5-year overall survival is <10%. Treatment options may include transarterial chemoembolization (TACE) and radiofrequency ablation. In terms of systemic treatment, the multi-kinase inhibitor sorafenib is the only first-line agent for HCC, but it only extends survival by 2.8 months in late-stage patients (7). Regorafenib (a multi-kinase inhibitor) (8) and nivolumab (a PD-1 inhibitor) (9) were recently approved as second-line treatment in patients who progressed on sorafenib, bringing the total number of approved systemic agents to three, still far fewer than most other cancer types.
Hepatocarcinogenesis is a complex, multi-step process of histologic transformation of normal hepatocytes to HCC, involving the accumulation of epi/genetic alterations in hepatocytes (10). In this review, we summarized some of the challenges in the diagnosis and clinical management of HCC, assessed the current status of circulating cell-free DNA (cfDNA) as a “liquid biopsy” in HCC, and discussed the outlook of how circulating cfDNA may address some of the challenges in the clinic.
Clinical Challenges in Liver Cancer
In the context of precision medicine, one of the biggest challenges in HCC clinical management is the frequent lack of tumor tissue for molecular profiling. Tumor materials may be obtained during resection and/or transplantation. However, these procedures are restricted to patients with early-stage, limited HCC (11). Unlike most other solid tumor types, the diagnosis of HCC does not always require the histopathologic analysis of tumor tissue. This is particularly true for patients with a known background of cirrhosis. In cirrhotic patients, HCC is frequently diagnosed on radiology alone or with serum alpha-fetoprotein (AFP) levels (11). Consequently, tumor materials for genetic profiling are unavailable for many unresectable HCC patients. Indeed, molecular profiling studies of HCC tumor tissues are frequently biased toward resectable tumors (12–23). Moreover, biopsy and surgical intervention are invasive procedures, and thus longitudinal sampling for the purpose of disease monitoring is clinically not feasible nor warranted in nearly all cases. An alternative source of DNA for molecular profiling would be highly desirable.
Early detection from screening programs has been reported to confer a survival benefit in HCC patients (24, 25). Currently, the most widely used blood-based biomarker is serum AFP. The clinical utility of AFP for diagnosis, even in the screening setting of high-risk patients, is limited by its lack of sensitivity and specificity. In fact, a substantial proportion of HCC patients do not display elevated serum AFP levels, while patients with chronic liver disease may also show elevated levels (26). Indeed, AFP is not currently recommended as the sole diagnostic marker (11).
Like virtually all solid tumors, variable levels of intra-tumor genetic heterogeneity with branched and parallel evolutionary patterns have been detected in HCC patients (27, 28). Genetic diversity appears to show spatial organization, with intrahepatic metastases showing rapid diversification at the distant site (28). HCC may also metastasize to distant organs, most frequently to the lungs, the lymph nodes, bone, and the adrenal glands. To add to the complexity, recurrent or multifocal HCC may represent intrahepatic metastasis, in which the malignant cells are disseminated from a single primary tumor or may represent independent (multi-centric) tumors (29). Clinical distinction between these two entities is important as intrahepatic metastases are likely to be more poorly differentiated and aggressive. Molecular analysis based on whole-genome sequencing in a recent study of 20 patients with synchronous or metachronous disease within the liver found that 65% (13/20) patients had evidence of multi-centric tumors (29). The direct clinical implication of intra-tumor genetic heterogeneity and multi-centricity is that a single biopsy may be inadequate to give a representative portrait of the epi/genomic landscape of the disease.
What is Circulating cfDNA?
Under the general nomenclature of “liquid biopsy,” the two main types are circulating tumor cells (CTCs) and cfDNA (Figure 1). CTCs refer to the intact tumor cells released into circulation. Analysis of CTCs offers the opportunity to study their behavior in experimental systems and to study them on the DNA, RNA, and protein levels (30). However, current methods detect only a few CTCs in a single blood draw of 7.5 ml (31, 32), limiting their clinical utility.
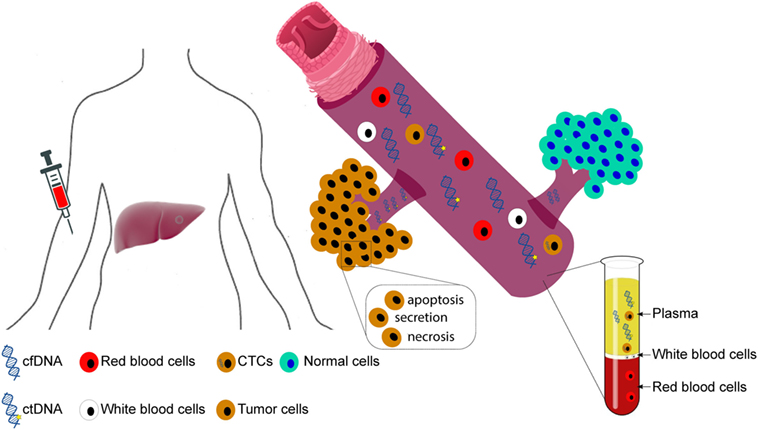
Figure 1. Circulating cell-free DNA (cfDNA) and circulating tumor cells (CTCs). Figure illustrates the origin of circulating cfDNA and CTCs. Circulating cfDNA may be released by apoptotic and necrotic cells, as well as through the secretion of living cells. In cancer patients, the fraction of tumor-derived cfDNA comprises circulating tumor DNA (ctDNA).
By contrast, circulating tumor DNA (ctDNA), the fraction of cfDNA derived from tumor cells, appears to be more readily detectable than CTCs (33). Most circulating cfDNA fragments are 160–180 base pair fragments, roughly the size of a mononucleosomal unit, suggesting that they are largely released by apoptotic cells (34, 35). In most cases, white blood cells are the biggest contributor of cfDNA in the plasma (36). By contrast, ctDNA should in theory harbor the same epi/genetic alterations as the originating tumor cells. Interestingly, HCC patients have been reported to have aberrantly shorter and longer cfDNA fragments, with the shorter fragments likely to have derived from tumor cells, while the longer fragments are hypothesized to have derived from necrotic cells (35).
Studies of the potential clinical utility of cfDNA are based on the hypothesis that abnormal forms of circulating cfDNA are more likely to be present in cancer patients. Both plasma-derived and serum-derived cfDNA/ctDNA have been and are being investigated as potential blood-based biomarkers for diagnosis, prognosis, and as a surrogate for molecular profiling in HCC (Table 1).
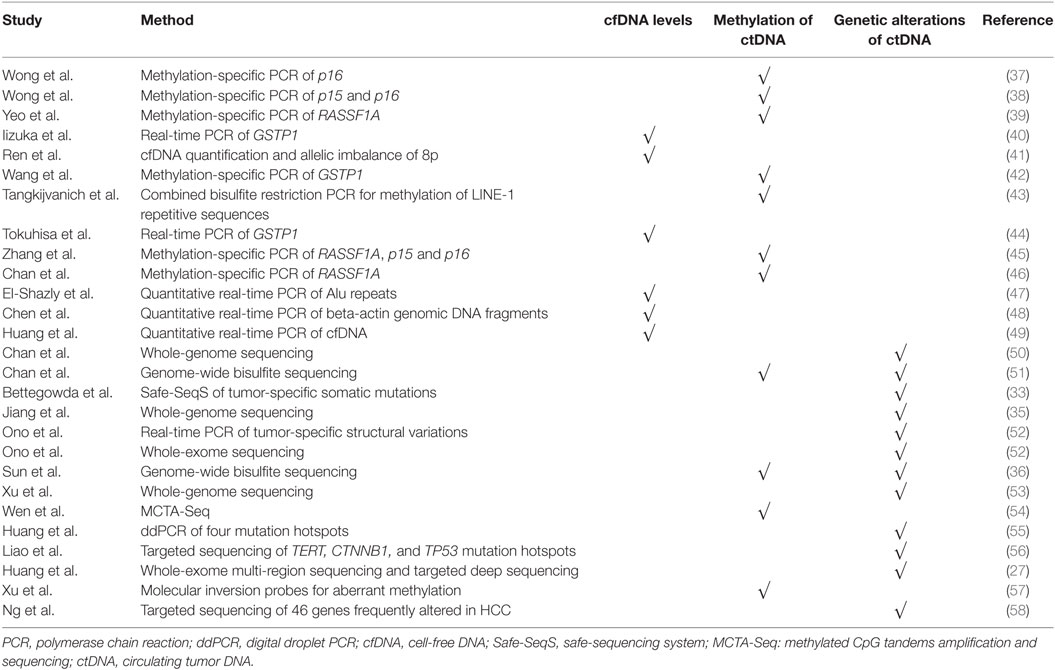
Table 1. List of studies reporting on the analysis of circulating cfDNA in HCC patients.
Quantitative Analysis of cfDNA in Hepatocellular Carcinoma
The initial description of elevated cfDNA concentration in cancer patients (59, 60) led the scientific community to further investigate the clinical utility of cfDNA concentration. In HCC, one of the first studies evaluating serum cfDNA concentrations in 52 HCV-associated HCC patients found that HCC patients had increased cfDNA concentrations compared to HCV carriers without known HCC and HCV-negative non-HCC individuals (40) (Table 1). The authors confirmed their findings in a follow-up study with a larger cohort of 96 HCV-associated HCC patients and 100 HCV carriers without known HCC (44). Since then, studies have reported the concentration of cfDNA in serum or plasma to be 3–4 times higher in HCC patients compared to patients with chronic hepatitis (44, 49, 61) and nearly 20 times that of healthy controls (49). Several studies, including our own, have also reported that high cfDNA concentration was associated with larger tumors (40, 41, 58), higher tumor grade (40, 58), shorter overall survival (41, 44), and may serve as predictive biomarker for distant metastasis after curative surgery (44).
Attempts to use cfDNA quantification as a diagnostic tool have been more mixed, with one study reporting that cfDNA detection using quantitative PCR could discriminate HCC from normal controls with 90% sensitivity and 90% specificity (49). However, a meta-analysis found that cfDNA alone lacked robustness as the sole diagnostic tool for HCC, but its performance could be enhanced when used in conjunction with serum AFP levels (62). Importantly, quantitative analysis of cfDNA, as opposed to ctDNA, does not provide information into the biological characteristics and potential molecular targets of the HCC. Distinguishing ctDNA from cfDNA would require alternative approaches such as genomic and/or epigenomic analyses focusing on tumor-specific alterations.
Methylation Analysis of ctDNA in Hepatocellular Carcinoma
DNA methylation is a common heritable mark of epigenetic regulation in eukaryotic organisms involving the covalent transfer of a methyl group to the C-5 position of the cytosine ring of DNA by DNA methyltransferases (63). Epigenetic mechanisms have been shown to play a pivotal role in the carcinogenesis of human tumors (64). It has been demonstrated that aberrant methylation of CpG islands serves as an important mechanism for the inactivation of tumor suppressor genes involved in hepatocarcinogenesis (42, 65–67).
The first studies to use methylation status as a tumor-specific marker to detect ctDNA in HCC patients investigated the methylation status of the p15 and p16 genes (37, 38) (Table 1), both of which are frequently abrogated in human neoplasms (68, 69). In these studies, the authors found that p15 and p16 were methylated in 16% (4/25) and 59% (13/22), respectively, of the plasma/serum DNA (37, 38). Importantly, the authors demonstrated that all cases with evidence of p15/16 methylation in the plasma/serum DNA also showed evidence of methylation in the corresponding tumors (37, 38), suggesting that ctDNA reflects the epigenetic status of the originating tumors. On the other hand, not all HCC tumors with p15/p16 methylation were associated with the equivalent methylation status in the plasma/serum DNA, underscoring the observation that ctDNA likely only accounts for a subset of cfDNA (36). Subsequent studies expanded the investigations to hypermethylation of the glutathione S-transferase P1 (GSTP1) and ras association domain family 1A (RASSF1A) genes (39, 42, 45, 46) and hypomethylation of LINE-1 repetitive sequence (43). While hypermethylation of GSTP1 was detected in 50% (16/32) of the cfDNA (42), it appears that RASSF1A methylation was a more sensitive marker, with 70–93% of the sera of HCC patients showing evidence of hypermethylation (45, 46). It has also been reported that aberrant methylation in ctDNA may identify AFP-negative HCC (46). In agreement with the quantification of cfDNA, methylation studies have found that aberrant methylation is associated with increased risk of metastasis or recurrence (38), larger tumors (39, 43), and worse prognosis (43, 46).
More recent studies have taken advantage of NGS-based genome-wide survey of the methylation landscape to identify diagnostic and prognostic methylation markers suitable for ctDNA profiling. One such study described the use of methylated CpG tandems amplification and sequencing for the genome-wide detection of hypermethylated CpG islands in the cfDNA of HCC patients (54). The authors identified TGS10, ST8SIA6, RUNX2, and VIM as the best hypermethylated markers for the detection of small HCC (<3 cm) (54). Another study used The Cancer Genome Atlas methylation profiles of HCC tumors and an independent data set of normal blood leukocytes to construct a diagnostic prediction model using 10 methylation markers (57). When tested in the cfDNA, the diagnostic models created by these studies achieved 94% sensitivity and 89% specificity in distinguishing HCC patients from cirrhotic or normal controls (54) and >83% sensitivity and >90% specificity in distinguishing HCC patients from normal controls (57). Both studies detected aberrant methylation in a subset of or all AFP-negative HCC (54, 57). One of the studies also showed that the diagnostic model could also differentiate HCC patients from those with liver diseases such as HBV/HCV infection and fatty liver disease and that the model scores correlated with tumor burden, treatment response, and disease stage (57).
On a more global scale, genome-wide hypomethylation is frequently observed in human cancer (51, 70). Genome-wide bisulfite sequencing of plasma-derived cfDNA showed that plasma methylation profiles among healthy individuals are fairly stable and did not show evidence of global hypomethylation (51). By contrast, plasma-derived cfDNA from predominantly HBV-positive and Barcelona Clinic Liver Cancer (BCLC) stage A HCC patients showed that a median of 34.1% (IQR 2.5–56.7%) of the genome was hypomethylated (51). These results illustrate that global hypomethylation associated with HCC is reflected in the cfDNA (51). Furthermore, cfDNA global hypomethylation as a marker to distinguish HCC patients from healthy individuals was reported to be 81% sensitive and 93% specific (51). Deconvolution of the methylation profiles suggests that while the liver contributes to ~10% of cfDNA in healthy individuals, the fraction rises to 24% (IQR 19.0–44.0%) in HCC patients (36). The variability among HCC patients is consistent with the substantial inter-individual variability in the proportion of hypomethylated genome (51), as it would be expected that the sensitivity to detect hypomethylation would be reduced in HCC patients with low fraction of ctDNA.
Besides being investigated as a diagnostic aid, methylation levels may also serve as a disease monitoring tool. For instance, rising serum concentrations of methylated RASSF1A have been reported in HBV carriers progressing to HCC (46). On the other hand, while it appears that methylation markers in the cfDNA are linked to the tumor, it is unclear whether they are derived directly from HCC tissue (54).
Genetic Analysis of ctDNA in Hepatocellular Carcinoma
Genomic characterization of HCC to the base pair resolution has revealed that no two HCCs harbor the same repertoire of somatic genetic alterations (13, 15, 16, 21–23). However, the somatic genetic alterations frequently found in HCC converge onto several main pathways, namely, p53 signaling (e.g., TP53 and CDKN2A), Wnt/β-catenin pathway (e.g., CTNNB1 and AXIN1), chromatin remodeling (e.g., ARID1A, ARID1B, ARID2, BAP1, MLL, MLL3, and PBRM1), response to oxidative stress (e.g., KEAP1 and NFE2L2), telomerase maintenance (e.g., TERT promoter mutation, HBV integration into TERT, TERT fusion genes, and TERT copy number amplification), among others (21, 23). With therapeutic decisions being increasingly genotype-based, a number of studies have investigated the detection of ctDNA on the basis of somatic genetic alterations in HCC.
The proportion of HCC patients with detectable ctDNA based on somatic genetic alterations varies wildly between studies. Using direct sequencing, the TP53 R249S mutation could be detected in 26% of the plasma DNA in an African HCC patient cohort (71). Using NGS investigating hotspot mutations in TERT promoter, CTNNB1, and TP53, tumor-associated mutations were detected in 8/41 (20%) plasma samples in a predominantly HBV-positive cohort (56) (Table 1). Interrogating TP53 c.747G>T (p.R249S), CTNNB1 c.121A>G (p.T41A) and c.133T>C (p.S45A), and TERT c.-124C>T promoter mutations using digital droplet PCR (ddPCR), ctDNA could be detected in 27/48 (56%) of the predominantly HBV-positive and BCLC stage A patients (55). Furthermore, copy number analysis found that 13/31 (42%) of HCC patients had detectable copy number alterations in the plasma (53). In the seminal study by Bettegowda et al., it was reported that the fraction of advanced stage HCC patients with at least one detectable somatic mutation in the plasma was 75% (3/4), which was similar to other solid tumors of non-brain origin (33).
The variability in the proportion of HCC patients with detectable ctDNA appears to be related to cohort composition and geographical locations, as well as the methodologies for detection. Nonetheless, consistent with cfDNA quantification and methylation studies, detectable ctDNA on the basis of tumor-specific genetic alterations is associated with well-established clinicopathologic parameters such as tumor size, AFP, and vascular invasion (52, 53, 56), as well as recurrence and extrahepatic metastasis (52, 56). Our own experience is largely consistent with these observations. In a recently published study of 30 HCC patients, we demonstrated that somatic mutations in genes frequently genetically altered were detected in 27% (8/30) of patients and in 86% (6/7) of patients with large tumor (≥5 cm diameter) or metastatic disease (58). We also observed that detectable ctDNA on the basis of tumor-specific mutations was positively correlated with tumor size and Edmondson grade, although not with BCLC stage or AFP levels (58).
The fraction of ctDNA in cfDNA, estimated based on the fraction of sequences harboring tumor-associated mutations, also appears to vary substantially between patients. Among the patients with detectable ctDNA, ddPCR of four hotspot mutations in TP53, CTNNB1, and TERT promoter found that the mutant allele fraction ranged from 0.33 to 23.7% (55). Our own NGS-based study identified mutant allele fractions from 0.06 to 45% in the cfDNA among patients with detectable ctDNA (58). Based on copy number alterations and/or methylation, it was estimated that a median of 24.0% (range: 4.3–71.4%) of cfDNA was tumor-derived (36, 50). However, even between three HCC patients with advanced stage disease, there was enormous variability, with 7.2, 15, and 7,910 mutant fragments detected per 5 ml of plasma (33).
Analysis of tumor-specific genetic alterations has also allowed us to determine how well ctDNA would reflect the biology of the tumors. Indeed, whole-genome sequencing performed in the plasma-derived cfDNA suggests that the copy number profiles of plasma DNA highly resemble those of the matched primary tumor (35, 50). Furthermore, hotspot mutations detected in the plasma DNA were almost always associated with the detection of the same mutations in the corresponding tumors (56). In our own study, comparing plasma-derived cfDNA and synchronously collected frozen biopsies from the primary tumor, we found that 87% (80/92) of the mutations were captured in the cfDNA among the seven patients in whom the largest tumor was ≥5 cm or was associated with metastasis (58). Importantly, we found that the proportion of mutations captured in the matched primary tumors of these patients was similar (95%, 87/92). Our results suggest that ctDNA reflects the biology of the primary tumors in patients with high disease burden. These findings are particularly important in the context of HCC, given that patients with high disease are the least likely to undergo surgical resection.
The more exciting recent development is the potential for ctDNA to circumvent the intra-tumor genetic heterogeneity in HCC (27, 28). Its potential was demonstrated with the use of whole-genome sequencing of the cfDNA from four HCC patients before surgery. The analysis revealed that 15–94% tumor-associated single nucleotide variants could be detected in the cfDNA, even at the very limited depth of 17× (50). Similarly, whole-exome sequencing of a patient with combined HCC/cholangiocarcinoma found that 63% of the tumor-associated mutations were observed in the serum DNA (52). However, the tumor and the serum samples for this patient were collected 2 years apart, which makes it difficult to assess whether the differences reflect the clonal evolution that may have occurred through two rounds of TACE (52). It may be argued that a synchronously collected cfDNA sample would have been more representative of the tumor. Indeed, synchronously collected cfDNA samples were representative of the primary tumors, but only in patients with high disease burden (58). We also found instances where mutations with low variant allele fraction (therefore almost certainly subclonal) in the primary tumors were readily detectable only in the cfDNA (58), suggesting that there is potential for ctDNA to circumvent the intra-tumor genetic heterogeneity.
The most comprehensive study to date to address the question whether cfDNA captures intra-tumor heterogeneity was conducted by multi-region whole-exome sequencing of five patients (27). The authors found that the cfDNA captured most of the mutations that were heterogeneous between tumor regions, but only given the knowledge of the mutations present in the tumor (27). On the other hand, agnostic to the mutations in the tumor, the authors concluded that cfDNA was insufficient in capturing the heterogeneity of the disease but could be used to identify actionable mutations that were ubiquitous in the tumor (27).
Given the apparent correlation between ctDNA fraction and disease burden (33, 58), genomic profiling is also being investigated as a disease monitoring tool and for the detection of recurrence prior to clinical recurrence. Similar to cfDNA levels and methylation analysis of ctDNA, quantification of ctDNA on the basis of somatic genetic alterations showed that the ctDNA level mirrors response to treatment and disease progression, in that the ctDNA level dropped or completely disappeared after resection and rose prior to recurrence or metastasis (50–52). By contrast, in patients that remained in long-term remission, ctDNA was not detectable in long-term follow-up cfDNA samples (51, 52).
Outlook: Addressing Clinical Problems in HCC
The characterization of ctDNA in HCC patients has focused on three main aspects: its potential for risk prediction or early cancer detection in a screening setting, for response to treatment and recurrence monitoring, and as a surrogate for tumor molecular profiling (Figure 2). With serum AFP levels being the only widely used but insensitive blood-based biomarker for HCC, a more sensitive and specific biomarker is highly desirable. While cfDNA level correlates with the disease stage and is easy to measure, it alone also lacks sensitivity and specificity (62). Moreover, elevated cfDNA level is not unique to HCC, as cfDNA level is also elevated in individuals with diabetes, stroke, acute trauma, pregnancy, among others (72).
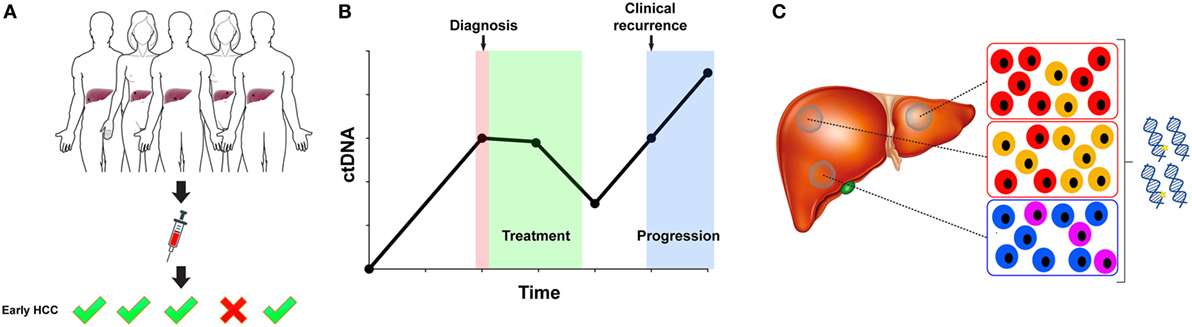
Figure 2. Circulating tumor DNA (ctDNA) for early detection, disease monitoring, and molecular profiling. (A) ctDNA is being investigated as a marker for the detection of early hepatocellular carcinoma (HCC). A suitable marker for early detection in the screening setting would require excellent specificity. (B) The fluctuation of ctDNA level may be informative in assessing response to treatment and in detecting minimal residual disease. (C) ctDNA profiling may circumvent sampling biases resulting from intra-tumor genetic heterogeneity and multi-centric HCC.
Despite the increased sensitivity in detection methods, establishing the utility of ctDNA for risk prediction or early cancer detection in a screening setting remains challenging. Overcoming the challenges posed by the low fraction of ctDNA within cfDNA in early-stage HCC would be crucial. Thus far, a study of 50 HCC patients with repeated serum DNA sampling suggested that aberrant methylation could be detected up to 9 years prior to the diagnosis of HCC (45). There are a number of other anecdotal examples to suggest that the detection of epi/genetic alterations in ctDNA may predate clinical diagnosis. For instance, whole-genome sequencing of two cases without known HCC revealed the presence of copy number alterations in the plasma cfDNA, and the patients were diagnosed with HCC 3–4 months later (35). Large case–control studies required to establish the utility of ctDNA in early cancer detection are inherently difficult to carry out, as they will have to be prospective, will involve repeated sampling at regular intervals, and with long-term follow-up of the patients.
Developing a ctDNA test for early cancer detection would involve the selection of an appropriate marker broadly applicable for a given cohort. Highly recurrent hotspot mutations such as TP53 R249S, CTNNB1 amino acids D32, S33, S37, T41, and S45, and TERT c.-124C>T promoter mutations (13, 15, 16, 21–23) may serve as potential markers for HCC detection. However, the discovery of frequent TERT promoter mutation in cirrhotic pre-neoplastic lesions (73) suggests that it may lack specificity in a screening setting. An intriguing possibility could be to detect ctDNA on the basis of shorter fragment sizes (35), although it is currently unclear whether shorter fragment size is a universal feature of ctDNA.
Compared to early detection, ctDNA for response and recurrence monitoring is closer to clinical application. In other cancer types, ctDNA is being used to detect minimal residual disease (74) and to track the emergence of drug resistant clones (75). HCC patients often undergo repeated rounds of therapeutic interventions such as TACE and ablation, with recurrence occurring several years later. Clonal evolution of the disease means that the genetic information obtained from tumor tissue at diagnosis or resection may become outdated by the time of recurrence. ctDNA profiling makes longitudinal monitoring of clonal evolution possible and may assist in the detection of micrometastatic disease (30). However, it should be noted that the timing of sample collection is an important consideration in the interpretation of the analysis, as it has been shown that serum ctDNA peaks 4 weeks after TACE (52).
Circulating tumor DNA profiling holds promise as a surrogate to overcome the challenges posed by intra-tumor genetic heterogeneity. It has been hypothesized that the detection of TERT promoter, CTNNB1, and TP53 mutations in the plasma cfDNA but not in the HCC tumor may be attributed to intra-tumor genetic heterogeneity (55, 56). However, similar to the challenges faced in early detection, the ability to capture all somatic genetic alterations is hampered by the sensitivity of current technologies. For instance, whole-exome sequencing of a single HCC patient suggests that 63% of the mutations were shared between the resected tumor and the serum cfDNA 2 years post-surgery, but only 27% of the mutations would have been detected had the samples been analyzed independently (52). Similar observations were made in a multi-region whole-exome sequencing study of five HCC patients (27), highlighting the technical challenges in using ctDNA as a surrogate for molecular profiling in the absence of tumor tissue. However, it should be noted that the patients included in the studies had resectable disease. Given the correlation of ctDNA and tumor burden, it is conceivable that a larger fraction of alterations would be represented and detectable in the ctDNA in late-stage patients. Indeed, we found that cfDNA profiling nearly recapitulated the repertoire of mutations detected in the primary tumors of patients with high disease burden (58), suggesting that the use of cfDNA for genetic profiling when biopsy is unavailable may be feasible in this subset of patients.
Currently, there are no widely accepted predictive markers for response to sorafenib or regorafenib. Response to immunotherapy such as nivolumab has been associated with tumor mutational burden (76). In fact, microsatellite instability status has recently been approved as an indication for nivolumab treatment of metastatic colorectal cancer (76). A subset of HCC exhibits a hypermutator phenotype (12, 21). With nivolumab approved as a second-line treatment, assessing mutational burden in the ctDNA of patients with advanced HCC should be feasible and may provide a predictive marker for response. Among our cohort of 30 patients, one HCC biopsy displayed a hypermutator phenotype, and the phenotype was clearly observed in the cfDNA (58). In addition, molecular analyses of ctDNA may uncover ubiquitous (i.e., present in all cancer cells) targetable genetic alterations that may quality patients for experimental therapy or off-label use. In a cohort of 70 HCC patients, 39% (27/70) harbored potential targetable alterations, including TSC1/TSC2 mutations (mTOR inhibitors), EGFR mutations (gefitinib and erlotinib), CCND1 amplifications and CDKN2A deletions or mutations (palbociclib), ATM mutation (olaparib), and MET amplification (tivantinib) (27). Using a more restricted targeted sequencing consisting of 46 coding and non-coding genes frequently altered in HCCs, we also detected a TSC2 frameshift mutation in the cfDNA and primary tumor of a metastatic patient (58).
Thus far, cfDNA profiling of HCC has focused on blood-based analysis (plasma and serum). It is plausible that ctDNA may also be detected in other body fluids, such as urine. Indeed, a recent study reported the use of qPCR-based analysis of TP53 c.747G>T (p.R249S) and aberrant methylation of the GSTP1 and RASSF1A genes in the urine of 10 HCC patients to monitor recurrence during follow-up (77). Of the five patients who were confirmed by MRI for recurrence, all had detectable changes in the urine cfDNA. Similar to plasma-derived cfDNA, the authors reported up to 9 months of lead-time prior to radiological recurrence (77). Compared to blood, urine is even more readily available. Further investigations into the use of urine would certainly be of clinical interest.
Through the analysis of cfDNA quantity, methylation, and genomic studies of ctDNA, it is clear that ctDNA holds potential to address a number of outstanding questions in the clinical management of HCC. Studies carried out thus far have laid the foundation for future studies into the use of ctDNA in early detection, response assessment, and detection of minimal residual disease. Exploiting ctDNA profiling to overcome the challenges posed by intra-tumor genetic heterogeneity may also provide biological insights into hepatocarcinogenesis.
Author Contributions
CN and SP conceived the study. CN, GDC, LT, and SP performed literature screening. CN and SP wrote the manuscript. All the authors reviewed and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Krebsliga Beider Basel (KLbB-4183-03-2017 to CKYN) and Krebsliga Schweiz (KFS-3995-08-2016 to SP). SP was funded by Swiss National Science Foundation (Ambizione grant number PZ00P3_168165); CN was supported by Swiss National Science Foundation (Ambizione grant number PZ00P3_168165) and by the European Research Council Synergy grant 609883 (MERiC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136:E359–86. doi:10.1002/ijc.29210
2. Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol (2006) 24:2137–50. doi:10.1200/JCO.2005.05.2308
3. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology (2007) 132:2557–76. doi:10.1053/j.gastro.2007.04.061
4. Grewal P, Viswanathen VA. Liver cancer and alcohol. Clin Liver Dis (2012) 16:839–50. doi:10.1016/j.cld.2012.08.011
5. Mair RD, Valenzuela A, Ha NB, Ayoub WS, Daugherty T, Lutchman GA, et al. Incidence of hepatocellular carcinoma among US patients with cirrhosis of viral or nonviral etiologies. Clin Gastroenterol Hepatol (2012) 10:1412–7. doi:10.1016/j.cgh.2012.08.011
6. El-Serag HB. Hepatocellular carcinoma. N Engl J Med (2011) 365:1118–27. doi:10.1056/NEJMra1001683
7. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med (2008) 359:378–90. doi:10.1056/NEJMoa0708857
8. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2017) 389:56–66. doi:10.1016/S0140-6736(16)32453-9
9. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (2017) 389:2492–502. doi:10.1016/S0140-6736(17)31046-2
10. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer (2006) 6:674–87. doi:10.1038/nrc1934
11. Bruix J, Sherman M; American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: an update. Hepatology (2011) 53:1020–2. doi:10.1002/hep.24199
12. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet (2012) 44:760–4. doi:10.1038/ng.2291
13. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet (2012) 44:694–8. doi:10.1038/ng.2256
14. Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology (2013) 58:1693–702. doi:10.1002/hep.26540
15. Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res (2013) 23:1422–33. doi:10.1101/gr.154492.113
16. Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM, Sung CO, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology (2014) 60:1972–82. doi:10.1002/hep.27198
17. Jhunjhunwala S, Jiang Z, Stawiski EW, Gnad F, Liu J, Mayba O, et al. Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol (2014) 15:436. doi:10.1186/s13059-014-0436-9
18. Shiraishi Y, Fujimoto A, Furuta M, Tanaka H, Chiba K, Boroevich KA, et al. Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers. PLoS One (2014) 9:e114263. doi:10.1371/journal.pone.0114263
19. Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet (2014) 46:1267–73. doi:10.1038/ng.3126
20. Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet (2014) 46:1160–5. doi:10.1038/ng.3101
21. Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet (2015) 47:505–11. doi:10.1038/ng.3252
22. Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet (2016) 48:500–9. doi:10.1038/ng.3547
23. Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell (2017) 169:1327–41.e1323. doi:10.1016/j.cell.2017.05.046
24. Zhang BH, Yang BH, Tang ZY. Randomized controlled trial of screening for hepatocellular carcinoma. J Cancer Res Clin Oncol (2004) 130:417–22. doi:10.1007/s00432-004-0552-0
25. van Meer S, De Man RA, Coenraad MJ, Sprengers D, Van Nieuwkerk KM, Klumpen HJ, et al. Surveillance for hepatocellular carcinoma is associated with increased survival: results from a large cohort in the Netherlands. J Hepatol (2015) 63:1156–63. doi:10.1016/j.jhep.2015.06.012
26. Lok AS, Sterling RK, Everhart JE, Wright EC, Hoefs JC, Di Bisceglie AM, et al. Des-gamma-carboxy prothrombin and alpha-fetoprotein as biomarkers for the early detection of hepatocellular carcinoma. Gastroenterology (2010) 138:493–502. doi:10.1053/j.gastro.2009.10.031
27. Huang A, Zhao X, Yang XR, Li FQ, Zhou XL, Wu K, et al. Circumventing intratumoral heterogeneity to identify potential therapeutic targets in hepatocellular carcinoma. J Hepatol (2017) 67:293–301. doi:10.1016/j.jhep.2017.03.005
28. Zhai W, Lim TK, Zhang T, Phang ST, Tiang Z, Guan P, et al. The spatial organization of intra-tumour heterogeneity and evolutionary trajectories of metastases in hepatocellular carcinoma. Nat Commun (2017) 8:4565. doi:10.1038/ncomms14565
29. Furuta M, Ueno M, Fujimoto A, Hayami S, Yasukawa S, Kojima F, et al. Whole genome sequencing discriminates hepatocellular carcinoma with intrahepatic metastasis from multi-centric tumors. J Hepatol (2017) 66:363–73. doi:10.1016/j.jhep.2016.09.021
30. Bardelli A, Pantel K. Liquid biopsies, what we do not know (Yet). Cancer Cell (2017) 31:172–9. doi:10.1016/j.ccell.2017.01.002
31. Kelley RK, Magbanua MJ, Butler TM, Collisson EA, Hwang J, Sidiropoulos N, et al. Circulating tumor cells in hepatocellular carcinoma: a pilot study of detection, enumeration, and next-generation sequencing in cases and controls. BMC Cancer (2015) 15:206. doi:10.1186/s12885-015-1195-z
32. Kowalik A, Kowalewska M, Gozdz S. Current approaches for avoiding the limitations of circulating tumor cells detection methods—implications for diagnosis and treatment of patients with solid tumors. Transl Res (2017) 185:58–84.e15. doi:10.1016/j.trsl.2017.04.002
33. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med (2014) 6:224ra224. doi:10.1126/scitranslmed.3007094
34. Jung K, Fleischhacker M, Rabien A. Cell-free DNA in the blood as a solid tumor biomarker – a critical appraisal of the literature. Clin Chim Acta (2010) 411:1611–24. doi:10.1016/j.cca.2010.07.032
35. Jiang P, Chan CW, Chan KC, Cheng SH, Wong J, Wong VW, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci U S A (2015) 112:E1317–25. doi:10.1073/pnas.1500076112
36. Sun K, Jiang P, Chan KC, Wong J, Cheng YK, Liang RH, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci U S A (2015) 112:E5503–12. doi:10.1073/pnas.1508736112
37. Wong IH, Lo YM, Zhang J, Liew CT, Ng MH, Wong N, et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res (1999) 59:71–3.
38. Wong IH, Lo YM, Yeo W, Lau WY, Johnson PJ. Frequent p15 promoter methylation in tumor and peripheral blood from hepatocellular carcinoma patients. Clin Cancer Res (2000) 6:3516–21.
39. Yeo W, Wong N, Wong WL, Lai PB, Zhong S, Johnson PJ. High frequency of promoter hypermethylation of RASSF1A in tumor and plasma of patients with hepatocellular carcinoma. Liver Int (2005) 25:266–72. doi:10.1111/j.1478-3231.2005.01084.x
40. Iizuka N, Sakaida I, Moribe T, Fujita N, Miura T, Stark M, et al. Elevated levels of circulating cell-free DNA in the blood of patients with hepatitis C virus-associated hepatocellular carcinoma. Anticancer Res (2006) 26:4713–9.
41. Ren N, Qin LX, Tu H, Liu YK, Zhang BH, Tang ZY. The prognostic value of circulating plasma DNA level and its allelic imbalance on chromosome 8p in patients with hepatocellular carcinoma. J Cancer Res Clin Oncol (2006) 132:399–407. doi:10.1007/s00432-005-0049-5
42. Wang J, Qin Y, Li B, Sun Z, Yang B. Detection of aberrant promoter methylation of GSTP1 in the tumor and serum of Chinese human primary hepatocellular carcinoma patients. Clin Biochem (2006) 39:344–8. doi:10.1016/j.clinbiochem.2006.01.008
43. Tangkijvanich P, Hourpai N, Rattanatanyong P, Wisedopas N, Mahachai V, Mutirangura A. Serum LINE-1 hypomethylation as a potential prognostic marker for hepatocellular carcinoma. Clin Chim Acta (2007) 379:127–33. doi:10.1016/j.cca.2006.12.029
44. Tokuhisa Y, Iizuka N, Sakaida I, Moribe T, Fujita N, Miura T, et al. Circulating cell-free DNA as a predictive marker for distant metastasis of hepatitis C virus-related hepatocellular carcinoma. Br J Cancer (2007) 97:1399–403. doi:10.1038/sj.bjc.6604034
45. Zhang YJ, Wu HC, Shen J, Ahsan H, Tsai WY, Yang HI, et al. Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA. Clin Cancer Res (2007) 13:2378–84. doi:10.1158/1078-0432.CCR-06-1900
46. Chan KC, Lai PB, Mok TS, Chan HL, Ding C, Yeung SW, et al. Quantitative analysis of circulating methylated DNA as a biomarker for hepatocellular carcinoma. Clin Chem (2008) 54:1528–36. doi:10.1373/clinchem.2008.104653
47. El-Shazly SF, Eid MA, El-Sourogy HA, Attia GF, Ezzat SA. Evaluation of serum DNA integrity as a screening and prognostic tool in patients with hepatitis C virus-related hepatocellular carcinoma. Int J Biol Markers (2010) 25:79–86.
48. Chen H, Sun LY, Zheng HQ, Zhang QF, Jin XM. Total serum DNA and DNA integrity: diagnostic value in patients with hepatitis B virus-related hepatocellular carcinoma. Pathology (2012) 44:318–24. doi:10.1097/PAT.0b013e328353a24c
49. Huang Z, Hua D, Hu Y, Cheng Z, Zhou X, Xie Q, et al. Quantitation of plasma circulating DNA using quantitative PCR for the detection of hepatocellular carcinoma. Pathol Oncol Res (2012) 18:271–6. doi:10.1007/s12253-011-9438-z
50. Chan KC, Jiang P, Zheng YW, Liao GJ, Sun H, Wong J, et al. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem (2013) 59:211–24. doi:10.1373/clinchem.2012.196014
51. Chan KC, Jiang P, Chan CW, Sun K, Wong J, Hui EP, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc Natl Acad Sci U S A (2013) 110:18761–8. doi:10.1073/pnas.1313995110
52. Ono A, Fujimoto A, Yamamoto Y, Akamatsu S, Hiraga N, Imamura M, et al. Circulating tumor DNA analysis for liver cancers and its usefulness as a liquid biopsy. Cell Mol Gastroenterol Hepatol (2015) 1:516–34. doi:10.1016/j.jcmgh.2015.06.009
53. Xu H, Zhu X, Xu Z, Hu Y, Bo S, Xing T, et al. Non-invasive analysis of genomic copy number variation in patients with hepatocellular carcinoma by next generation DNA sequencing. J Cancer (2015) 6:247–53. doi:10.7150/jca.10747
54. Wen L, Li J, Guo H, Liu X, Zheng S, Zhang D, et al. Genome-scale detection of hypermethylated CpG islands in circulating cell-free DNA of hepatocellular carcinoma patients. Cell Res (2015) 25:1376. doi:10.1038/cr.2015.141
55. Huang A, Zhang X, Zhou SL, Cao Y, Huang XW, Fan J, et al. Detecting circulating tumor DNA in hepatocellular carcinoma patients using droplet digital PCR is feasible and reflects intratumoral heterogeneity. J Cancer (2016) 7:1907–14. doi:10.7150/jca.15823
56. Liao W, Yang H, Xu H, Wang Y, Ge P, Ren J, et al. Noninvasive detection of tumor-associated mutations from circulating cell-free DNA in hepatocellular carcinoma patients by targeted deep sequencing. Oncotarget (2016) 7:40481–90. doi:10.18632/oncotarget.9629
57. Xu RH, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater (2017) 16:1155–61. doi:10.1038/nmat4997
58. Ng CKY, Di Costanzo GG, Tosti N, Paradiso V, Coto-Llerena M, Roscigno G, et al. Genetic profiling using plasma-derived cell-free DNA in therapy-naïve hepatocellular carcinoma patients: a pilot study. Ann Oncol (2018):mdy083. doi:10.1093/annonc/mdy083
59. Leon SA, Green A, Yaros MJ, Shapiro B. Radioimmunoassay for nanogram quantities of DNA. J Immunol Methods (1975) 9:157–64. doi:10.1016/0022-1759(75)90106-4
60. Stroun M, Anker P, Lyautey J, Lederrey C, Maurice PA. Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol (1987) 23:707–12. doi:10.1016/0277-5379(87)90266-5
61. Yang YJ, Chen H, Huang P, Li CH, Dong ZH, Hou YL. Quantification of plasma hTERT DNA in hepatocellular carcinoma patients by quantitative fluorescent polymerase chain reaction. Clin Invest Med (2011) 34:E238. doi:10.25011/cim.v34i4.15366
62. Liao W, Mao Y, Ge P, Yang H, Xu H, Lu X, et al. Value of quantitative and qualitative analyses of circulating cell-free DNA as diagnostic tools for hepatocellular carcinoma: a meta-analysis. Medicine (Baltimore) (2015) 94:e722. doi:10.1097/MD.0000000000000722
63. Robertson KD. DNA methylation and human disease. Nat Rev Genet (2005) 6:597–610. doi:10.1038/nrg1655
64. Sceusi EL, Loose DS, Wray CJ. Clinical implications of DNA methylation in hepatocellular carcinoma. HPB (Oxford) (2011) 13:369–76. doi:10.1111/j.1477-2574.2011.00303.x
65. Tchou JC, Lin X, Freije D, Isaacs WB, Brooks JD, Rashid A, et al. GSTP1 CpG island DNA hypermethylation in hepatocellular carcinomas. Int J Oncol (2000) 16:663–76. doi:10.3892/ijo.16.4.663
66. Zhong S, Tang MW, Yeo W, Liu C, Lo YM, Johnson PJ. Silencing of GSTP1 gene by CpG island DNA hypermethylation in HBV-associated hepatocellular carcinomas. Clin Cancer Res (2002) 8:1087–92.
67. Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol (2003) 163:1101–7. doi:10.1016/S0002-9440(10)63469-4
68. Cairns P, Mao L, Merlo A, Lee DJ, Schwab D, Eby Y, et al. Rates of p16 (MTS1) mutations in primary tumors with 9p loss. Science (1994) 265:415–7. doi:10.1126/science.8023167
69. Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature (1994) 368:753–6. doi:10.1038/368753a0
70. Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature (1983) 301:89–92. doi:10.1038/301089a0
71. Kirk GD, Camus-Randon AM, Mendy M, Goedert JJ, Merle P, Trepo C, et al. Ser-249 p53 mutations in plasma DNA of patients with hepatocellular carcinoma from The Gambia. J Natl Cancer Inst (2000) 92:148–53. doi:10.1093/jnci/92.2.148
72. Swarup V, Rajeswari MR. Circulating (cell-free) nucleic acids – a promising, non-invasive tool for early detection of several human diseases. FEBS Lett (2007) 581:795–9. doi:10.1016/j.febslet.2007.01.051
73. Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun (2013) 4:2218. doi:10.1038/ncomms3218
74. Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med (2015) 7:302ra133. doi:10.1126/scitranslmed.aab0021
75. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature (2013) 497:108–12. doi:10.1038/nature12065
76. Overman MJ, Mcdermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol (2017) 18:1182–91. doi:10.1016/S1470-2045(17)30422-9
Keywords: cell-free DNA, circulating tumor DNA, hepatocellular carcinoma, liquid biopsy, somatic mutations, copy number alterations, methylation
Citation: Ng CKY, Di Costanzo GG, Terracciano LM and Piscuoglio S (2018) Circulating Cell-Free DNA in Hepatocellular Carcinoma: Current Insights and Outlook. Front. Med. 5:78. doi: 10.3389/fmed.2018.00078
Received: 24 December 2017; Accepted: 08 March 2018;
Published: 26 March 2018
Edited by:
Venancio Avancini Alves, University of São Paulo, BrazilReviewed by:
Fernando Schmitt, Universidade do Porto, PortugalMarco Volante, Università degli Studi di Torino, Italy
Copyright: © 2018 Ng, Di Costanzo, Terracciano and Piscuoglio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charlotte K. Y. Ng, kiuyancharlotte.ng@usb.ch;
Salvatore Piscuoglio, salvatore.piscuoglio@usb.ch