 Xueling Wu1,2Xiaoyan Wu1Li Shen1,2
Xueling Wu1,2Xiaoyan Wu1Li Shen1,2 Jiaokun Li1,2Runlan Yu1,2Yuandong Liu1,2Guanzhou Qiu1,2
Jiaokun Li1,2Runlan Yu1,2Yuandong Liu1,2Guanzhou Qiu1,2 Weimin Zeng1,2*
Weimin Zeng1,2*- 1School of Minerals Processing and Bioengineering, Central South University, Changsha, China
- 2Key Laboratory of Biometallurgy, Ministry of Education, School of Minerals Processing and Bioengineering, Central South University, Changsha, China
Few bacteria are resistant to tetracycline and can even biodegrade tetracycline in the environment. In this study, we isolated a bacterium Pandoraea sp. XY-2, which could biodegrade 74% tetracycline at pH 7.0 and 30°C within 6 days. Thereafter, we determined the whole genome sequence of Pandoraea sp. XY-2 genome is a single circular chromosome of 5.06 Mb in size. Genomic annotation showed that two AA6 family members-encoding genes and nine glutathione S-transferase (GSTs)-encoding genes could be relevant to tetracycline biodegradation. In addition, the average nucleotide identities (ANI) analysis between the genomes of Pandoraea sp. XY-2 and other Pandoraea spp. revealed that Pandoraea sp. XY-2 belongs to a new species. Moreover, comparative genome analysis of 36 Pandoraea strains identified the pan and specific genes, numerous single nucleotide polymorphisms (SNPs), insertions, and deletion variations (InDels) and different syntenial relationships in the genome of Pandoraea sp. XY-2. Finally, the evolution and the origin analysis of genes related to tetracycline resistance revealed that the six tetA(48) genes and two specificgenes tetG and tetR in Pandoraea sp. XY-2 were acquired by horizontal gene transfer (HGT) events from sources related to Paraburkholderia, Burkholderia, Caballeronia, Salmonella, Vibrio, Proteobacteria, Pseudomonas, Acinetobacter, Flavimaricola, and some unidentified sources. As a new species, Pandoraea sp. XY-2 will be an excellent resource for the bioremediation of tetracycline-contaminated environment.
Introduction
Tetracycline represents a major proportion of the broad-spectrum antibiotics widely used in prevention and therapy of human and animal diseases and as promoters for animal growth (Huber, 1971; Mesgari Abbasi et al., 2011; Wan et al., 2013; Leong et al., 2016). Because antibiotics cannot be absorbed or metabolized completely by the animals, 30–90% of antibiotics are excreted into the environment via urine and feces as the unmetabolized parent compound (Bound and Voulvoulis, 2004). Residues of antibiotic has increased the frequency of horizontal gene transfer (HGT) and resistance gene fixation in genomes, leading to the development of diverse resistance genes in genomic islands (Gillings and Stokes, 2012). Residues of antibiotic in the environment could cause the emergence and development of antibiotic-resistant genes and bacteria, which brings profound negative impacts on the health of human and animals (Chee-Sanford et al., 2001; Daghrir and Drogui, 2013; Zhu et al., 2013). Compared with other conventional methods such as hydrolysis and photolysis, microbial degradation of tetracycline is considered to be an inexpensive, eco-friendly, and high efficient technology for the removal of tetracycline from the environment (Jiao et al., 2008; Migliore et al., 2012). In recent years, there are only a few tetracycline-degrading microorganisms that have been reported. Zhang et al. (2015) found that 57.8% tetracycline could be degraded by Advenella kashmirensi which was selected from sewage of a pharmaceutical factory. Leng et al. (2016) have isolated tetracycline-degrading bacteria S. maltophilia strain DT1 from sewage of a contaminated soil.
Most antibiotic-degrading bacteria are drug-resistance bacteria because they need to survive in the presence of antibiotics to play their role of degradation. There are three mechanisms of tetracycline resistance: a ribosomal protection mechanism, an efflux pump mechanism, and an enzymatic inactivation mechanism (Speer and Salyers, 1989; Thaker et al., 2010; Roberts et al., 2011). Leng et al. (2017) have found that ribosomal protection and efflux pump mechanism were presented in the tetracycline resistance strain Stenotrophomonas maltophilia DT1. Leski et al. (2013) found that various pathogens, such as Klebsiella pneumoniae and Bacteroides fragilis, contained a flavin-dependent monooxygenase encoded gene tet(X), which can inactivate tetracycline.
The third-generation sequencing platform PacBio RS II and annotation analysis of the whole genome data can offer increased read lengths, unbiased genome coverage and identify the genes involved in the xenobiotic biodegradation and drug-resistance (Ferrarini et al., 2013; Kim et al., 2015; Huang et al., 2018). Jung et al. (2012) have identified the genes related to pectin metabolism in pectin degrader Alishewanella aestuarii B11(T) based on genomic analysis. Moreover, comparative genomics analyses can reveal the molecular implications associated with genetic diversity affecting the evolutionary origin and how these genetic differences are generated among distinct lineages (Teng et al., 2016). In particular, the phylogeny, the genetic traits and the evolutionary history of specific resistance pathways can also be obtained through comparative genomic analysis (Wang et al., 2017). Comparative genomics analyses can shed light on the genetic basis underlying the degradation ability to organic contaminants of bacteria. Therefore, investigating the resistance mechanisms in the tetracycline resistant bacteria at the molecular level is important to predicting the environmental fate of antibiotics and assessing the biological impacts of antibiotics on the microbes in the environment.
The genus Pandoraea was first proposed in 2,000 following the isolation from the sputum of cystic fibrosis patients (Coenye et al., 2000). Today, a total of 10 species with valid nomenclature (Parte, 2014) in the genus Pandoraea have been described. Members of Pandoraea tend to exhibit broad resistance to ampicillin, extended-spectrum cephalosporins, aztreonam, aminoglycosides, and meropenem (Daneshvar et al., 2001; Stryjewski et al., 2003). To date, Pandoraea spp. have been considerably attractive for biotechnological applications with various degradation abilities such as lignin degradation, polychlorinated biphenyls (PCBs) biodegradation, and chlorobenzen degradation (Baptista et al., 2010; Dhindwal et al., 2011; Shi et al., 2013). In addition, Pandoraea spp. used for bioremediation were essentially isolated from the environment, such as rotten stumps, petroleum refinery waste, and municipal wastewater treatment plant (Sarkar et al., 2017; Fang et al., 2018; Yang et al., 2018). However, to our best knowledge, no documentation of tetracycline biodegradation in the genus of Pandoraea has ever been reported to date. Moreover, there are relatively few analyses on the comparative genomics of Pandoraea genus.
In the present study we report that a tetracycline-degrading bacterium, Pandoraea sp. XY-2, was isolated and its tetracycline biodegrading behavior under various time, temperature, and initial pH was investigated. To further investigate the genome structure and function of Pandoraea sp. XY-2, we performed whole genome sequencing and annotation on Pandoraea sp. XY-2 using the Pacific Biosciences RSII single-molecule real-time (SMRT) platform and other related functional annotation databases. We then calculated the average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH) values between the available genomes of the whole Pandoraea group in order to ascertain whether Pandoraea sp. XY-2 represented a new species within the Pandoraea group. Additionally, we constructed the genomic evolution events involving this strain and compared the gene and genomic structure with those of closely related Pandoraea species to assess the phylogenetic relationship, genetic diversity and genomic structure variation. Finally, the genetic traits and evolutionary history of genes related to tetracycline resistance in genus Pandoraea were investigated. The insights gained in this study provides information useful for further clarifying the molecular mechanisms behind tetracycline biodegradation and resistance.
Materials and Methods
Chemicals, Sampling, and Media
All chemicals were purchased from Sinopharm Chemical Reagent Co. Ltd., including tetracycline standard, tryptone, yeast extract, NaCl, glucose, chromatography grade methanol and acetonitrile, analytical grade sodium dihydrogen phosphate, oxalic acid, disodium ethylenediaminetetraacetate, and citric acid.
The soil sample was obtained from the sludge in the sediment pool of a pig factory using tetracycline antibiotics as feed additive for a long-term (4 a) in Changsha. The average temperature and pH value of the sampling location in this study was 20°C and about 6.8. The latitude and longitude of our sampling locations are 28.1932 and 112.6973, respectively.
Lysogeny broth (LB) medium contained the following (in g L−1): tryptone, 10; yeast extract, 5; NaCl, 5; glucose, 10. The pH of LB medium was adjusted to 7.0–7.2 before autoclaving at 121°C for 20 min.
Bacterial Strain Isolation and Identification
0.1 g soil sample was made into suspension with 10 mL sterile water. One milliliter supernatant was added to the LB medium (An additional volume of tetracycline stock solution was added to the culture medium so that the tetracycline concentration was 20 mg L−1). The mixed solution was incubated at 30°C on a dark shaker at 150 rpm. After 3 days, 0.5 mL of the solution was inoculated into the LB medium containing 40 mg L−1 tetracycline and the other conditions remained unchanged, and then cultured for 6 days. The above procedures were repeated until the final concentration of tetracycline reached 120 mg L−1. After the enrichment, the solution was serially diluted from 10−1 to 10−9, and then 0.5 mL of each diluted solution was added to petri dishes and daub uniformly. Six colonies with different morphology were obtained after multiple purifications, and then inoculated separately into LB medium for cultivation. Only the isolate with a high level of degradation efficiency of tetracycline was further analyzed. Finally, Pandoraea sp. XY-2 was identified through 16S rRNA sequence analysis as previously described (Mangwani et al., 2015).
Analytical Methods of Residual Tetracycline
Concentration of tetracycline residues were analyzed by high-performance liquid chromatography (HPLC). A C18 reversed-phase column (Agilent Technologies) was operated at 30°C, with a 0.5 mL min−1 mobile phase consisting of 10 mmol L−1 sodium dihydrogen phosphate solution and acetonitrile. The injection volume was 20 μL, and the column gradient elution was monitored by a UV detector at 360 nm. Five milliliter solution from each reactor was centrifuged at 8,000 rpm at 4°C for 15 min. Four milliliter supernatant was added to a centrifuge tube containing 4 mL Na2EDTA-Mcllvaine buffer, then 1 mL n-hexane and 1 mL chloroform were added, and the mixture was shaked for 2 min and sonicated for 30 s. After the mixture was centrifuged at 8,000 rpm for 15 min, the supernatant was filtered through a 0.22 μm microporous membrane filter and preserved at 4°C for further testing with the previously described chromatographic conditions.
Tetracycline Biodegradation Experiments
In order to obtain the optimal conditions for Pandoraea sp. XY-2 biodegrading tetracycline, we studied the growth of Pandoraea sp. XY-2 and the biodegradation percentage of tetracycline under various culture time (i.e., 1, 2, …, 8, 9 day), temperatures (i.e., 20, 25, 30, 35, 40°C) and initial pH values (i.e., 5.0, 6.0, 7.0, 8.0, 9.0). Only biodegradation was considered because hydrolysis of tetracycline could be ignored during the testing period (Supplementary Figure 1). In these experiments, the amount of each bottle of culture solution was 150 mL. Pandoraea sp. XY-2 culture solution was inoculated to LB medium containing 50 mg L−1 tetracycline at 3% inoculum concentration, and then placed on a dark shaker at 150 rpm. During the culture time tests, the temperature and initial pH were 30° C and 7.0, respectively. During the different temperature tests, the initial pH was 7.0. During the pH tests, the temperature was 30°C. The final growth of Pandoraea sp. XY-2 and the biodegradation percentage of tetracycline in temperature and pH tests were measured only at the 6th day. All experiments were performed in triplicates. The growth of Pandoraea sp. XY-2 was measured by a hemocytometer and the concentration of tetracycline in the medium was measured using HPLC.
DNA Extraction, Genome Sequencing, Assembly, and Annotation
Genomic DNA of Pandoraea sp. XY-2 was extracted using the Genomic DNA Extraction Kit (Tian Gen biochemical technology Co. Ltd., Beijing, China).
Genomic DNA was sheared in a Covaris g-TUBE, and DNA fragments were then damage repaired, terminal repaired and purified using AMpure PB magnetic beads. SMRTbell libraries prepared with DNA Template Kit 2.0 and quantified by Qubit concentration. Agilent 2,100 was used to detect the size of the inserted fragments. Then, sequencing carried out using the PacBio RS II SMRT sequencing technology (Beijing Nuohe Zhiyuan Technology Co., Ltd., Beijing, China).
De novo genome assembly of PacBio reads for XY-2 (91,372 reads totaling 1,072,300,065 bp, mean length 11,735 bp, N50 length 16,735 bp) was conducted using the hierarchical genome assembly process (HGAP3 protocol and SMRT Link v.5.0.1 software) (Rhoads and Au, 2015). The mapping of assembled contigs onto long reads from the PacBio platform was performed by BlasR (Chaisson and Tesler, 2012). After mapping, long reads with high accuracy were assembled by the Celera Assembler v8.0 (Science New York, 2000). Then, the assembly resulted in one polished contig, which was a circular chromosome with no gaps.
Gene prediction carried out by GeneMarkS software (http://topaz.gatech.edu/). The tRNAscan-SE v1.3.1 and rRNAmmer v1.2 were used for identifying tRNA and rRNA, respectively (Schattner et al., 2005; Lagesen et al., 2007). Additionally, online Web-based tool Island Viewer v.3 was used for detecting genomic islands (Dhillon et al., 2015). To predict Interspersed Repeats, RepeatMasker was used (Chen, 2004). Tandem Repeats were identified for by using Tandem Repeat Finder v.4.09 (Benson, 1999). Finally, the gene functions were further investigated by BLASTP using the following five different function databases: the carbohydrate-active enzymes (CAZy) database (http://www.cazy.org/), the non-redundant (NR) protein database (http://www.ncbi.nlm.nih.gov/protein), the clusters of orthologous groups (COG) database (https://www.ncbi.nlm.nih.gov/COG/), the GO database (http://www.geneontology.org/), and the KEGG database (http://www.genome.jp/kegg/pathway.html).
Average Nucleotide Identity (ANI) Calculations and Phylogenetic Analyses
Before ANI analysis, the 16S rRNA sequence of type strains of Pandoraea spp. were retrieved from the EzTaxon server (http://www.eztaxon.org/, Jongsik et al., 2007). Comparative sequence analysis of the 16S rRNA gene of Pandoraea sp. XY-2 agaist the type strains of Pandoraea spp. was implemented within the server. The phylogenetic tree of Pandoraea sp. XY-2 based on the 16SrRNA gene sequences was constructed using the NJ method in MEGA v7.0 with 1,000 bootstraps replications (Kumar et al., 2016).
Reference genomes for comparison purposes were retrieved from the GenBank database (http://www.ncbi.nlm.nih.gov/genbank/). Whole genome comparisons to determine the ANI between genome sequences were produced using JSpeciesws database (http://jspecies.ribohost.com/jspeciesws) that use Blast alignments to evaluate whole genome homologies. Cut-offs for species delineation were 95% ANI on 69% of conserved DNA according to Goris et al. (2007). In addition, we also calculate the dDDH values using Genome-to-Genome Distance Calculator (Alsaari et al., 2015). The 70% species cut-off of dDDH usually kept in taxonomic studies of bacteria (Konstantinidis and Tiedje, 2007).
The other 35 Pandoraea strains (Table 1) from NCBI database were selected to perform the phylogenetic analysis of Pandoraea sp. XY-2. Firstly, the single-copy core genes were identified by core-pan analysis between Pandoraea sp. XY-2 and the 35 Pandoraea strains. The number and total length of concatenated single copy genes were 1,903 and 27,797 characters. Then, the multiple sequences alignment of single-copy core genes was performed using MUSCLE software. Finally, phylogenetic tree was generated using the NJ method in Mega v7.0 with 1,000 bootstraps replications.
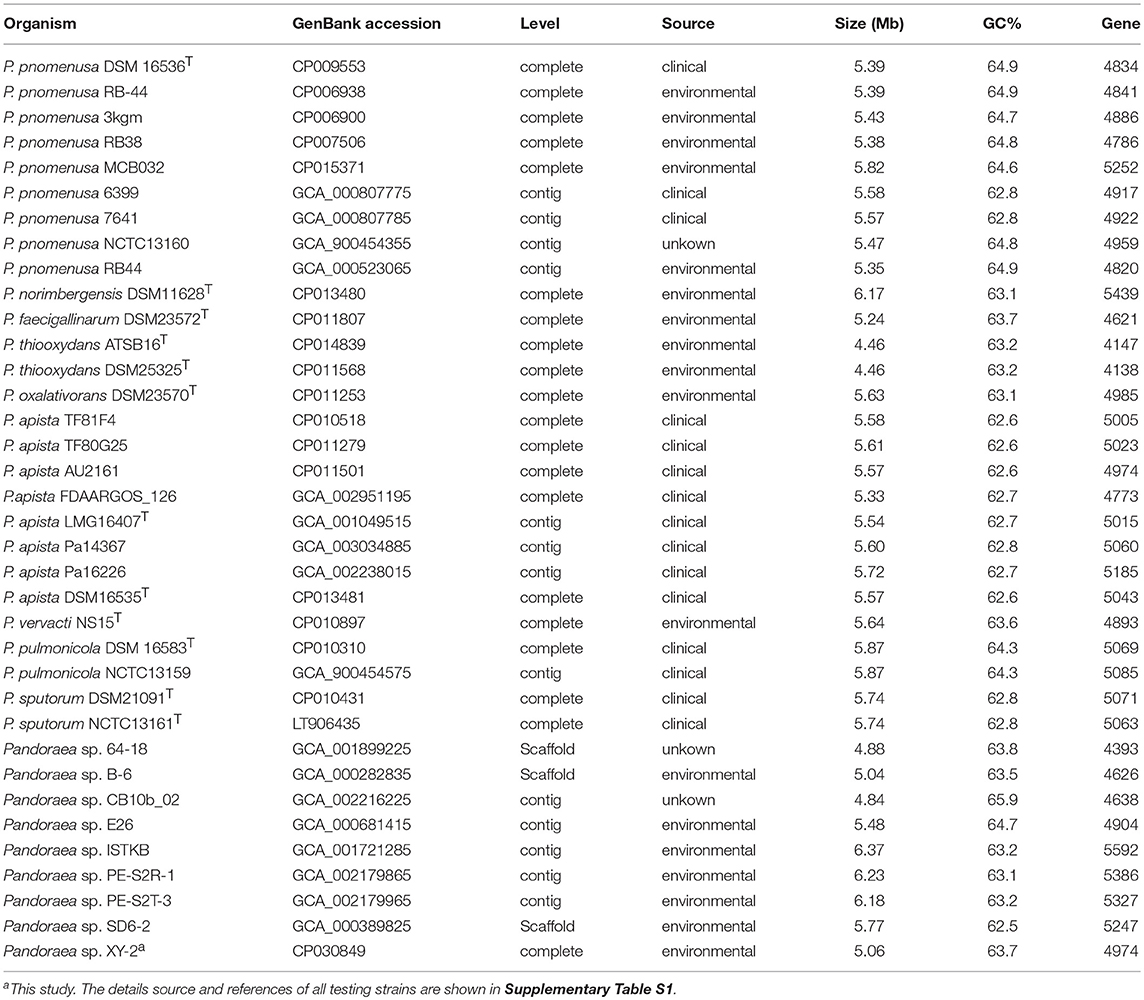
Table 1. Genomes statistical information of the 36 strains used in this study.
Comparative Genomics Analyses
We used the Bacterial Pan Genome Analysis tool (BPGA) pipeline (Chaudhari et al., 2016) to identify orthologous groups among Pandoraea testing genomes and to extrapolate the pan-genome models of applying default parameters. The statistics information and details source of all testing strains used are listed in Table 1 and Supplementary Table S1. We defined the set of genes shared among all strains as their core genome, the set of genes shared with more than two but not all strains as their accessory genome, the set of genes not shared with other strains in each strain as the specific genes and the set of non-homologous genes with all testing genomes as the pan genome. Finally, functional categories were assigned to gene clusters by the KEGG annotation previously described.
The size of Pandoraea pan-genome was extrapolated implementing a power law regression function Ps = κnγ using a built-in program of BPGA pipeline (Chaudhari et al., 2016), where Ps is the total number of non-orthologous gene families within its pan-genome, n is the number of tested strains, and both κ and γ are free parameters (Tettelin et al., 2008). The exponent γ < 0 suggests the pan-genome is “closed” considering the size of the pan-genome is reaching a constant as extra genomes adding in. Conversely, species is predicted to harbor an “open” pan-genome for 0 < γ < 1. Furthermore, the size of the core-genome was extrapolated fitting into an exponential decay function Fc = κcexp(−n/τc) + Ω, with a built-in program of BPGA pipeline (Chaudhari et al., 2016), where Fc is the number of core gene families, while κc, τc, and Ω are free parameters (Tettelin et al., 2005).
For variation analysis, we use P. apista DSM16535T as reference strain. The single nucleotide polymorphisms (SNPs) were identified by aligning scaffolds to reference using MUMmer 3.06 package (Kurtz et al., 2004). Reliable SNPs were obtained after using BLAST, TRF, and Repeatmask software to predict the repeat region of the reference sequence, and filter out the SNPs located in the repeat region. InDels (insertion and deletion variations) were detected through gap extension alignment between the whole genome de novo assembly and the reference using LASTZ software (Harris, 2007). Synteny analysis was performed using MUMmer v3.06 package.
Genes for Tetracycline Resistance
The coding sequences that are resistant to antibiotic was predicted using the Resistance Gene Identifier (RGI) software in the Comprehensive Antibiotic Resistance Database (CARD) (Mcarthur and Wright, 2015). After identifying the tetracycline resistance genes, each resistance gene was compared on NCBI database to find the genes with high similarity. Then, the evolutionary history of genes for tetracycline resistance was inferred using MEGA v7.0 (Kumar et al., 2016) with the Neighbor-Joining method with 1,000 bootstraps.
GenBank Accession Numbers
The sequence data of the whole genome of Pandoraea sp. XY-2 have been deposited to NCBI data base, with the accession number of CP030849.
Results
Isolation and Identification of Tetracycline-Degrading Bacteria
After enrichment, isolation, and purification, 6 strains named XY-1, XY-2, XY-3, XY-4, XY-5, and XY-6 were obtained. The colony morphology of strain XY-1 was round, white, opaque, while that of strain XY-3 was dry, white, uneven edges. The colony morphology of strain XY-4, XY-5, and XY-6 was cotton-like, snowflake and fluffy shape, respectively. All the strains were capable of biodegrade tetracycline, and the biodegrade percentage were between 36 and 65%. The strain XY-2 was selected for further analyses due to its highest ability to biodegrade tetracycline. The colony of strain XY-2 on LB solid medium was smooth and moist, and was light milky white with neat edges. It is a gram-negative bacterium and has a short rod shape under a scanning electron microscope (Supplementary Figure 2) and has a size of ~0.7 μm × (1.2–3) μm. After the 16S rDNA sequence analysis of strain XY-2, it was concluded that strain XY-2 belongs to genus Pandoraea in molecular phylogenetic classification and it was named Pandoraea sp. XY-2.
The Optimum of Biodegradation Parameters of Tetracycline by Pandoraea sp. XY-2
Figure 1 shows the effect of incubation time on the growth of Pandoraea sp. XY-2 and tetracycline biodegradation percentage. The growth of Pandoraea sp. XY-2 entered the logarithmic phase at the 3rd day, the biomass reached 2.1 × 1010 cfu/mL at the 6th day, and then the growth entered the stationary phase. The biodegradation percentage of tetracycline increased significantly during the first 6 days and it reached 73.8% at the 6th day. After that, there was almost no significant change with prolonged incubation.
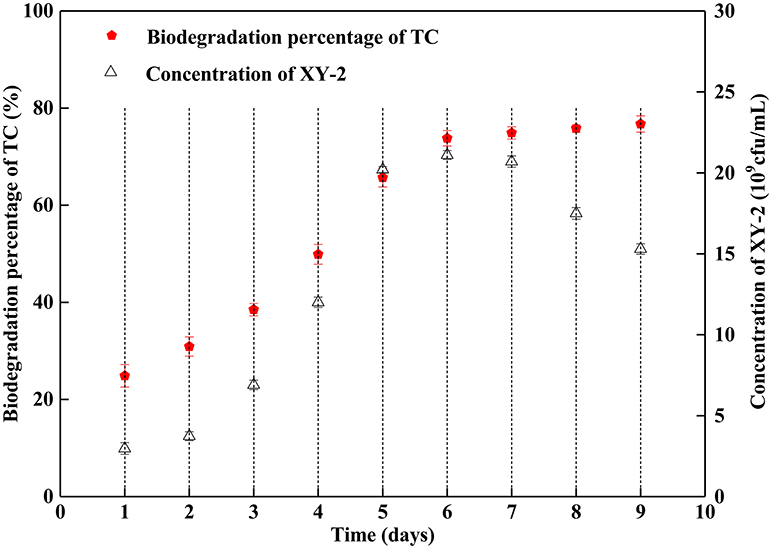
Figure 1. Effect of incubation time on the growth of Pandoraea sp. XY-2 and tetracycline biodegradation percentage. TC: tetracycline. XY-2: Pandoraea sp. XY-2. The initial concentration of tetracycline was 50 mg L−1. Pandoraea sp. XY-2 were cultured with an initial pH 7.0 at 30°C.
Effect of incubation temperature on the growth of Pandoraea sp. XY-2 and tetracycline biodegradation ability is shown in Figure 2. The growth of Pandoraea sp. XY-2 was greatly affected by temperature. With the increasing temperature from 30 to 40°C, the biomass of Pandoraea sp. XY-2 decreased to 1.57 × 1010 cfu/mL and the biodegradation percentage of tetracycline reached the maximum value and then remained stable at the 6th day.
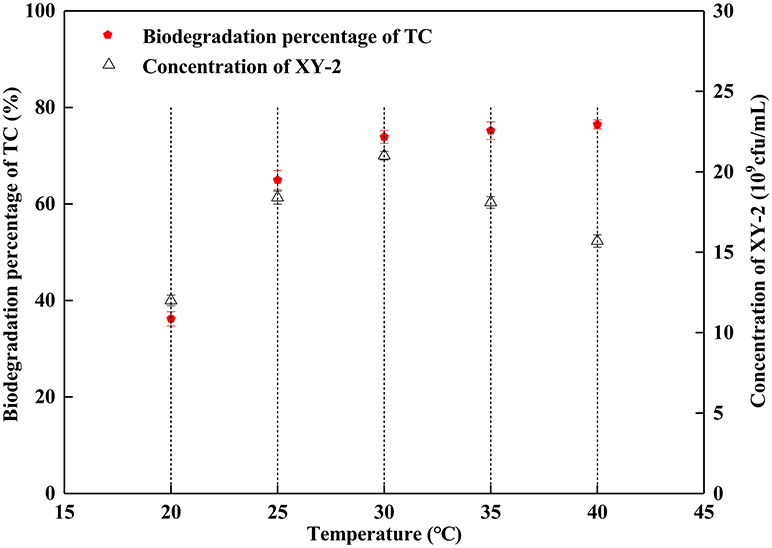
Figure 2. Effect of incubation temperature on the growth of Pandoraea sp. XY-2 and tetracycline biodegradation percentage. TC: tetracycline. XY-2: Pandoraea sp. XY-2. The effect of incubation temperature was tested at the 6th day. The initial concentration of tetracycline was 50 mg L−1. The initial pH was 7.0.
The proper pH value can affect not only the growth rate of microorganisms, but also the activity of enzymes. In this study, the growth of Pandoraea sp. XY-2 was significantly inhibited when the initial pH was 5 and 9 (Figure 3). The Pandoraea sp. XY-2 grew most vigorously and the biodegradation percentage of tetracycline reached the maximum value at pH 7. It was concluded that the growth of Pandoraea sp. XY-2 and tetracycline biodegradation percentage generally showed a trend of increasing first and decreasing then with the increasing initial pH.
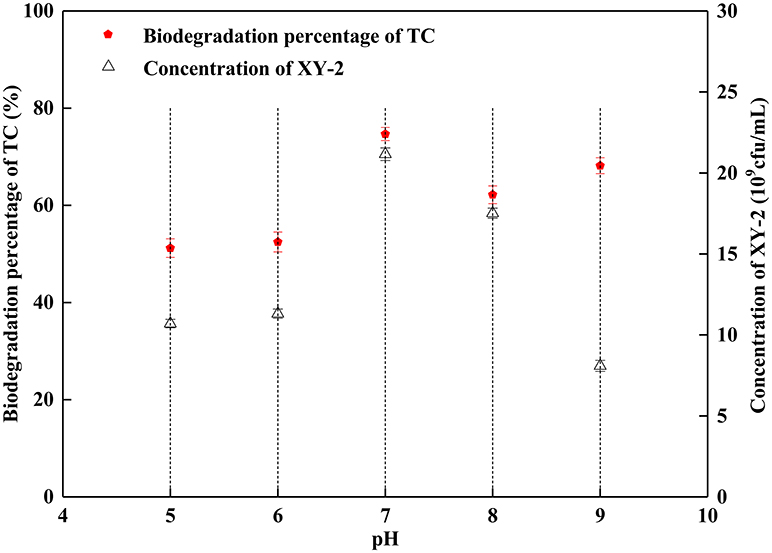
Figure 3. Effect of initial pH on the growth of Pandoraea sp. XY-2 and tetracycline biodegradation percentage. TC: tetracycline. XY-2: Pandoraea sp. XY-2. The effect of initial pH was tested at the 6th day. The initial concentration of tetracycline was 50 mg L−1. The temperature was 30°C.
Combined with the growth of Pandoraea sp. XY-2 and tetracycline biodegradation percentage, we selected a pH of 7, temperature of 30°C and culture period of 6 day as the optimum culture conditions for subsequent experiments.
Genome Characteristics and Functional Annotation of Pandoraea sp. XY-2
After sequencing, a total of 91,372 reads were obtained. Genome characteristics of the newly sequenced genome of Pandoraea sp. XY-2 are shown in Table 2. The Pandoraea sp. XY-2 has a single circular chromosome and the estimated genome size was 5,056,206 bp with a GC content of 63.76% (Figure 4). No plasmid was found in the genome sequence of this bacterium. The number of predicted genes was 4,974 with an estimated total length of 4,382,283 bp which makes up 86.67% of the entire genome. Out of all the predicted genes, 3,489 (70%) could be functionally annotated in the NCBI non-redundant (nr) database. A total of 1,485 hypothetical proteins were predicted for XY-2. There are 77 RNA genes consisting of 65 tRNA and 12 rRNA (4 5S rRNA, 4 16S rRNA, and 4 23S rRNA) genes. Eight GIs were detected in the genome of the Pandoraea sp. XY-2. Approximately 5,470 bp of interspersed repeats were found in the genome of the Pandoraea sp. XY-2, which accounted for 0.1082% of its genome size, whereas 27,270 bp of tandem repeats were found, which accounted for 0.5393% of its genome size. Tandem repeats were the predominant type of repeat region, accounting for 83.3% of the repeat regions in the Pandoraea sp. XY-2 genome.
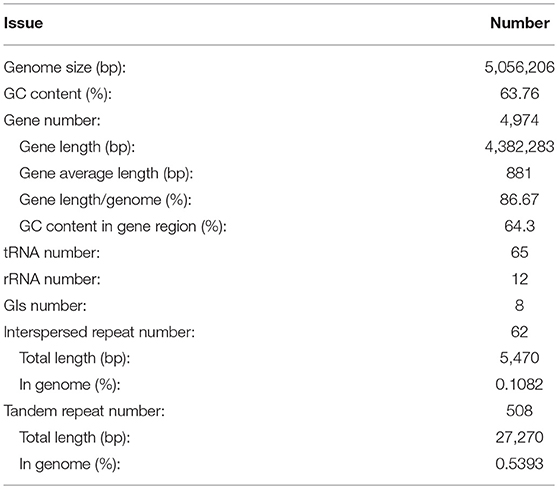
Table 2. Genome characteristics of the newly sequenced genome of Pandoraea sp. XY-2.
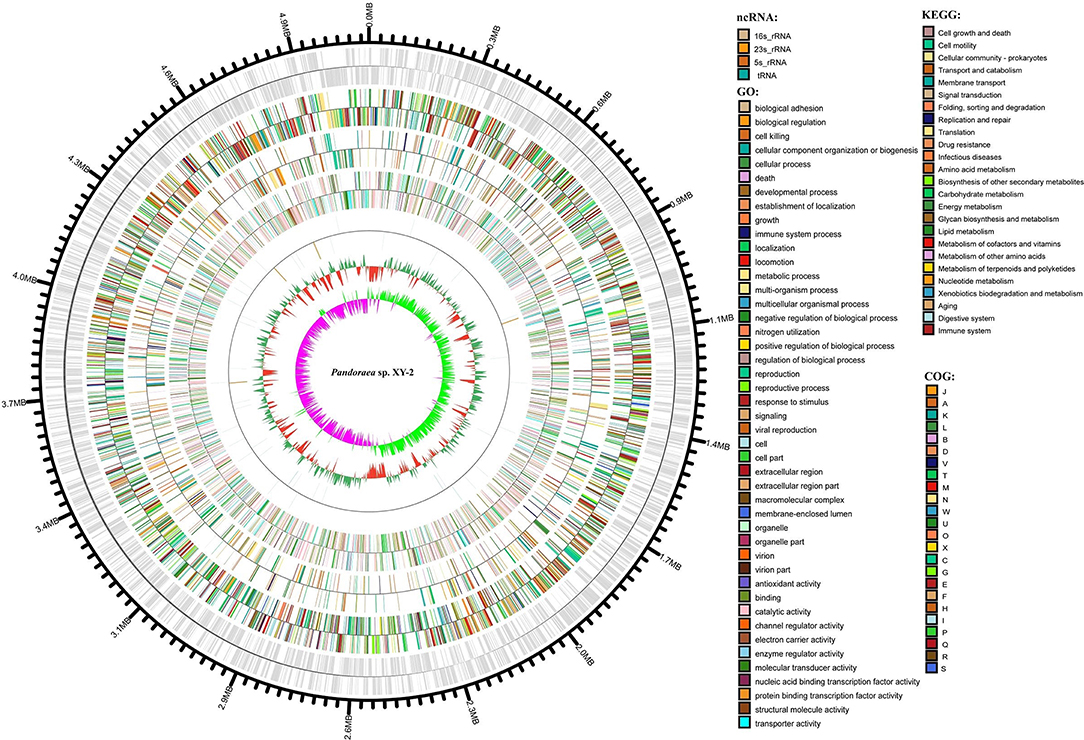
Figure 4. Circular representation of Pandoraea sp. XY-2. Based on, tRNA and rRNA, GO, KEGG, COG results, circular diagrams of chromosomes are drawn. The concentric circles show (reading outwards): GC skew, GC content, tRNA and rRNA, GO, KEGG, COG, genes, DNA coordinates.
Secreted CAZymes are crucial for bacteria biological activity. CAZymes are divided into five classes: glycoside hydrolases (GHs), carbohydrate esterases (CEs), polysaccharide lyases (PLs), glycosyltransferases (GTs), and auxiliary activities (AAs). Based on the CAZy annotation pipeline, we identified 65 CAZymes, including 5 AAs, 16 GHs, 30 GTs, 3 CEs, and 11 carbohydrate binding modules (CBMs) in the Pandoraea sp. XY-2 genome (Supplementary Figure 3). In the present study, CAZyme annotation revealed that Pandoraea sp. XY-2 contains a total of 2 families with 5 AAs in its genome sequence. Additionally, AA family classification also revealed that the components of AAs were 3 AA3 family members [glucose-methanol-choline (GMC) oxidoreductase, alcohol oxidase, aryl-alcohol oxidase/glucose oxidase, cellobiose dehydrogenase, pyranose oxidase), and 2 AA6 family members (1,4-benzoquinone reductase).
Clusters of orthologous groups (COG) database annotation showed that a number of genes are related to the basic functions of bacterial cells, including amino acid transport and metabolism (E), transcription (K), and energy production and conversion (C) (Supplementary Figure 4). Particularly, the number of genes participated in carbohydrate transport and metabolism (G), secondary metabolites biosynthesis, transport and catabolism (Q) and intracellular trafficking, secretion, and vesicular transport (U) were 244 (5.4%), 166 (3.6%), and 79 (1.7%), respectively. The GO annotation revealed that cellular processes and metabolic processes are very active in biological process; cell and cell part play a dominant role in cellular component; binding and catalytic activity play important roles in molecular function (Supplementary Figure 5). Furthermore, KEGG annotation showed that a total of 115 genes participated in the xenobiotic biodegradation and metabolism (Supplementary Figure 6 and Supplementary Table S2). It is worth noting that there were 5 genes encoding monooxygenase, 9 genes encoding dioxygenase, nine genes encoding glutathione S-transferase (GSTs) and 92 genes encoding other types of enzymes. Previous reports showed that tetracycline can be more readily biodegradable by using the detoxifying enzyme GSTs (Park and Choung, 2007). Accordingly, we selected all the 9 GSTs-encoding genes in Pandoraea sp. XY-2 together with GSTs-encoding genes from close-related strains for comparative analysis to shed light on the variation in the ability to degrade tetracycline between Pandoraea spp. The phylogenetic analysis of nine GSTs-encoding genes derived from Pandoraea sp. XY-2 and other close-related strains revealed that the GSTs-encoding genes were present in both environmental and clinical isolates and the evolution of these genes had no major variation (Supplementary Figure 7). We also calculated the GSTs-encoding genes contents in Pandoraea spp. and the results showed that the contents of these genes did not change significantly between environmental and clinical isolates (Supplementary Table S1).
ANI Calculations and Phylogenetic Analyses
The phylogenetic tree of Pandoraea sp. XY-2 based on 16SrRNA gene sequence was shown in Supplementary Figure 8. Pandoraea sp. XY-2 was most closely related to the P. apista LMG16407T (99.59 similarity). The 16S rRNA gene of Pandoraea sp. XY-2 having a similarity of 96.9–99.72% compared to the type strains of Pandoraea spp. (Supplementary Table S3). (Yarza et al., 2008) have reported that a new proposed genus would have ~6% divergence in 16S rRNA gene sequence from its closest genus. Therefore, Pandoraea sp. XY-2 was not a new genus. Stackebrandt and Ebers (2006) suggested that a 16S rRNA gene sequence similarity of 98.7–99% should become the boundary for delineation of prokaryotic species. Above this value, there is a need to prove by genome to genome comparisons whether distinct isolates belong to the same species. Accordingly, genomes comparisons of Pandoraea sp. XY-2 against all Pandoraea spp. were carry out for species circumscription.
ANI is considered to be the most relevant comparative parameter used for bacterial species delineation (Rosselló-Móra and Amann, 2015). Using ANI, the bacterial species delimitation borders can be set to about 94–96% identity, which would generally represent ~70% dDDH (Richter and Rossellómóra, 2009). Table 3 contain the matrix of nucleotide identities between whole genomes calculated under JSpeciesws database. The ANI% values of Pandoraea sp. XY-2 against all reference strains were < 85% (Table 3), which was a value far below the threshold of 94–96% identity that would serve as a boundary for species circumscription. Besides, the dDDH results showed that the dDDH% values of Pandoraea sp. XY-2 against all reference strains ranging from 14.9 to 52.2% (Supplementary Table S4).
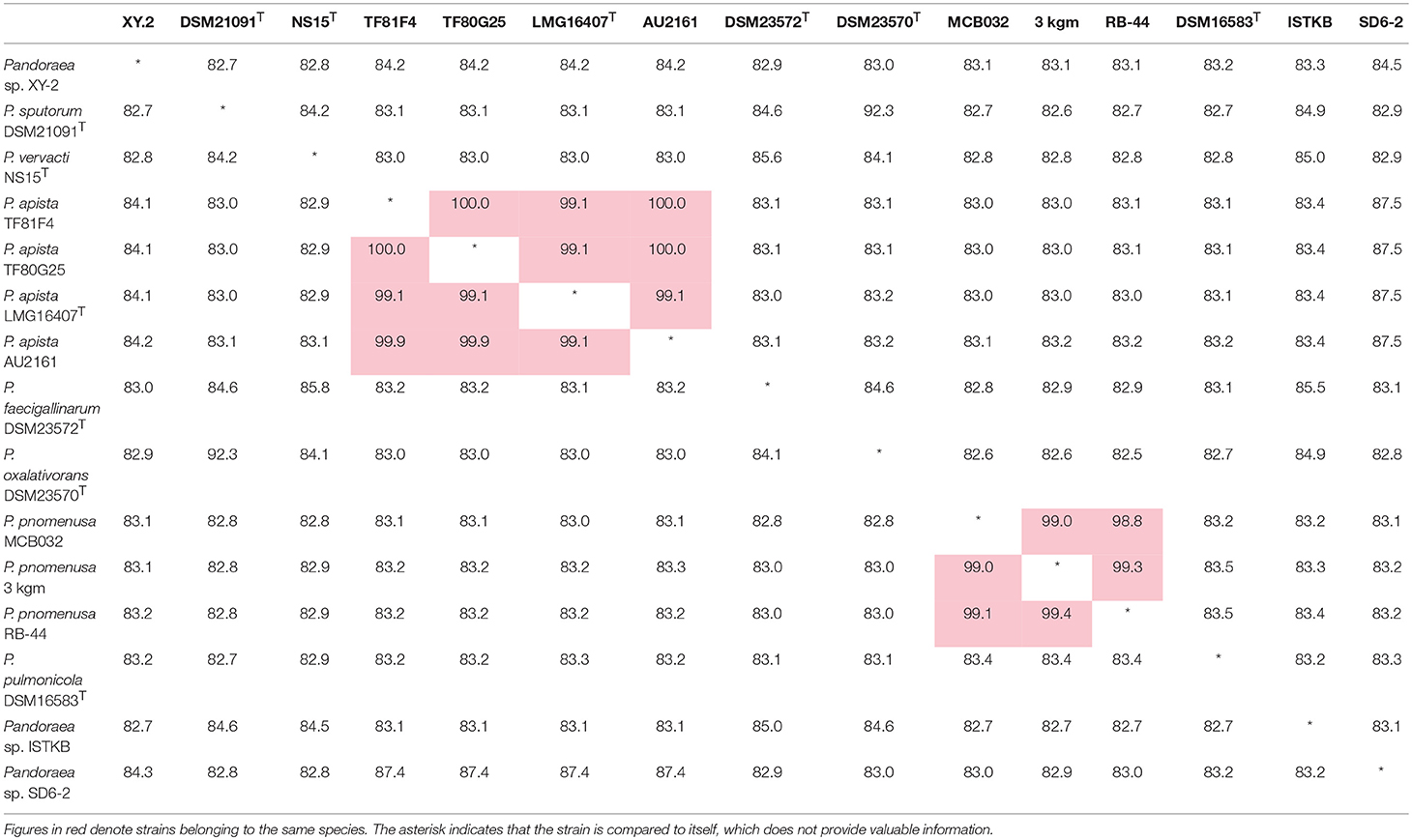
Table 3. Average nucleotide identities (ANI) analysis. Genome comparison of Pandoraea sp. XY-2 and other Pandoraea species.
A phylogenetic analysis based on the single-copy core genes was conducted to determine the relationship of Pandoraea sp. XY-2 with other 35 Pandoraea strains. Except for the sequencing data of Pandoraea sp. XY-2, other whole genomic data of 35 Pandoraea strains were downloaded from NCBI. The Pandoraea sp. XY-2 was clustered together with Pandoraea sp. 64–18 and Pandoraea sp. B-6 (Figure 5), which was indicative of intimate intragenus relationship among Pandoraea sp. XY-2, Pandoraea sp. 64–18 and Pandoraea sp. B-6. In addition, Pandoraea sp. XY-2 branches near the base of the Pandoraea sp. 64–18 and Pandoraea sp. B-6. We inferred that Pandoraea sp. XY-2 was likely arised before the Pandoraea sp. 64–18 and Pandoraea sp. B-6.
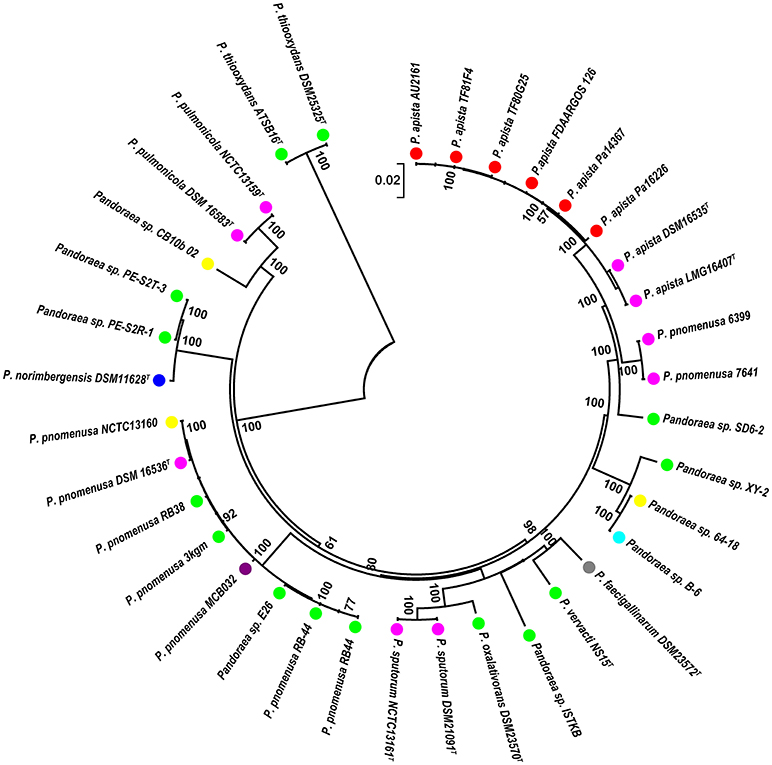
Figure 5. Phylogenetic analysis of Pandoraea sp. XY-2 based on all of the single-copy core genes among 36 whole genome sequences. The phylogeny tree was generated using the NJ method in Mega 7.0 with 1,000 bootstraps replications. The number and total length of concatenated single copy genes were 1,903 and 27,797 characters. The whole genome sequences of 35 other Pandoraea strains were retrieved from GenBank. The source of each strain was marked in different colors. Red, sputum; Pink, CF patient; Green, soil; Yellow, unknown; Blue, bamboo slips; Gray, oxalate-enriched culture; Purple, bioreactor; Navy blue, food.
Comparative Genomics Analyses
A pan-genome for the Pandoraea sp. XY-2 and 35 other Pandoraea strains was determined by PGAP pipeline. The total pan-genome for the 36 compared Pandoraea strains encompasses 186,995 putative protein-coding genes. Of these, 1,903 (1.02% of total pan-genome) were core conserved genes across all 36 strain genomes compared. Out of these 186,995 genes, 2,280 (1.22%) were represented in the accessory genomes of Pandoraea sp. XY-2. After comparing strain-specific genes, the Pandoraea sp. ISTKB had the highest amount of these (n = 899), while the isolate sequenced in this study had 414 specific genes. The number of specific genes in all 36 strains ranges from 2 to 899 (Figure 6). The KEGG annotation of specific genes of Pandoraea sp. XY-2 showed that a total of 10 genes involved in the environmental information processing, including one TetR family protein-encoding gene tetR (XY.2_GM004468) and one DHA1 family protein-encoding gene tetG (XY.2_GM004469) (Supplementary Figure 9).
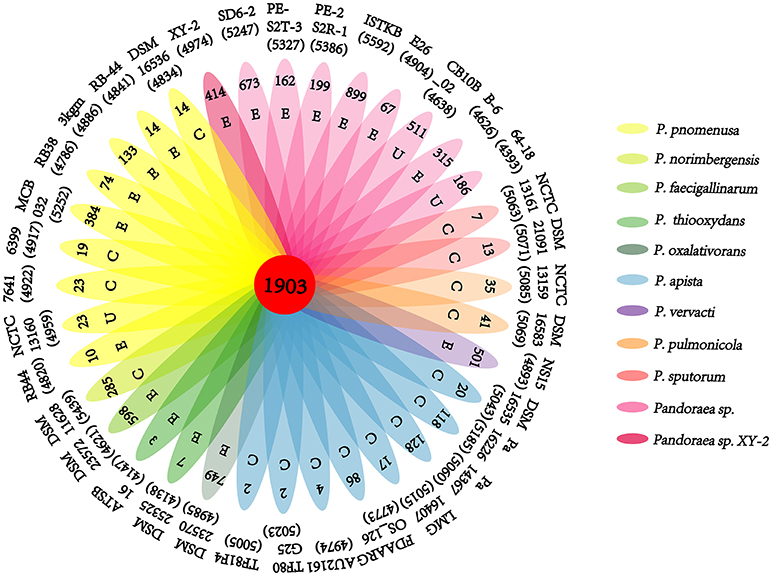
Figure 6. Pan-genome analysis of strains in the genus Pandoraea. Petal diagram of Pan-genome. The total number of protein coding genes within each genome is listed below the strain name. Numbers in non-overlapping portions of each oval show the number of CDSs specific to each strain. Letters in non-overlapping portions of each oval show the source of each strain (E, environment; C, clinical; U, unknown). The center is the number of orthologous coding sequences shared by all strains (i.e., the core genome).
Previous reports showed that mathematical extrapolation of pan-genome would be highly reliable provided that sufficient genomes (>5) are involved (Vernikos et al., 2015). The deduced power law regression function [Ps (n) = 3119.72 n0.50637] revealed that the pan-genome of Pandoraea had a parameter (γ) of 0.50637 falling into the range 0 < γ < 1, which indicating that the pan-genome was open (Figure 7) (Tettelin et al., 2008). In addition, the deduced exponential regression [Fc (n) = 3900.47e−0.0178922n] revealed that the extrapolated curve of core genome followed a steep slope, reaching a minimum of 1,903 gene families after the 36th genome was added (Figure 6).
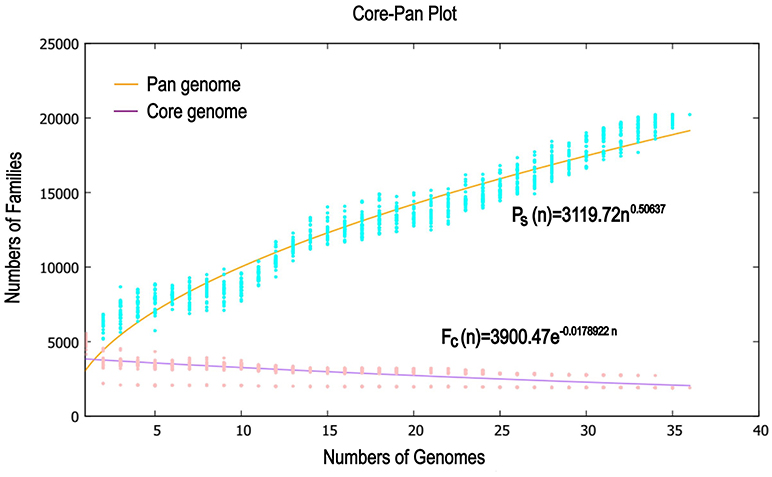
Figure 7. Mathematical modeling of pan genome and core genome of 36 Pandoraea strains.
The other 18 Pandoraea strains, which genome sequences level were complete, from NCBI database were selected to perform the phylogenetic analysis of Pandoraea sp. XY-2. The results showed that Pandoraea sp. XY-2 was clustered together with P. apista DSM16535T (Supplementary Figure 10). Combined with that it has not yet been reported that tetracycline could be biodegraded by Pandoraea spp., so we selected Pandoraea sp. XY-2 and P. apista DSM16535T to perform a genome-wide comparison. The genome-wide comparison identified all the SNPs, a total of 372,574 high-quality SNPs for Pandoraea sp. XY-2 were obtained (Supplementary Table S5). Most of the high-quality SNPs in the Pandoraea sp. XY-2 were located in CDS regions (333,149, 89.42%), with only 39,425 (10.58%) located in intergenic regions. A total of 67,476 non-synonymous SNPs and 264,974 synonymous SNPs were located within CDS regions, which resulted in a non-synonymous/synonymous (dN/dS) ratio of 0.255.
Gene gains and losses have been considered as one of the most significant contributors to functional changes (Nei and Rooney, 2005). We analyzed the insertion/deletion variations (InDels) between Pandoraea sp. XY-2 and P. apista DSM16535T and the results are shown in Supplementary Table S6. We identified 581 InDels in the chromosome of Pandoraea sp. XY-2 and the proportion of base deletion (60.76%) is higher than that of base insertion (39.24%). Most of the InDels were located in intergenic regions (388, 66.78%). Furthermore, synteny comparison of genomes between Pandoraea sp. XY-2 and P. apista DSM16535T showed that the two genomes have 550 collinear blocks covering 73.91 % of Pandoraea sp. XY-2 genome. On the other hand, some regions between Pandoraea sp. XY-2 and P. apista DSM16535T displays inversion, translocation, and Tran+Inver, revealing different syntenial relationships to the reference sequence (Figure 8).
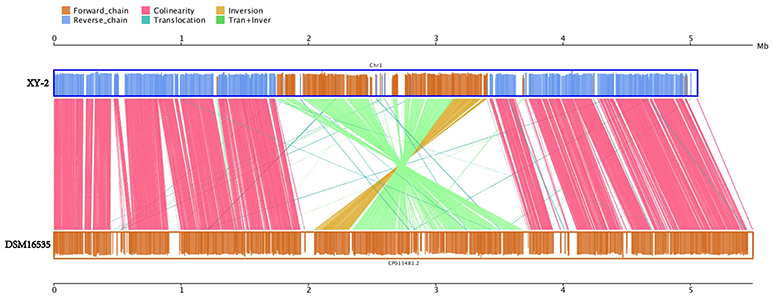
Figure 8. Synteny comparison of genomes between Pandoraea sp. XY-2 and P. apista DSM16535T. In the upper and lower axes, the brown frame represents the genome forward chain, the blue frame represents the genome reverse chain, the height of the color filled in the frame represents the similarity degree of the comparison. The color comparison type of the linked graph between the upper and lower axes: Collinear (red), Translocation (green), Inversion (yellow), and Tran+Inver (light green).
The Origin and Evolution of Tetracycline Resistance Genes
From CARD analysis, a total of 218 genes were identified as having a role in the resistance to various antibiotics, of which 9 were tetracycline resistance genes (Supplementary Table S7). The type and number of tetracycline resistance genes were 6 tetA(48), 1 tetB(P), 1 tetG, and 1 tetT (Supplementary Table S8). Resistance is primarily due to either energy-dependent efflux of tetracycline or protection of the ribosomes from the action of tetracycline. TetA(48) and tetG belong to the efflux pump gene, while tetB(P) and tetT belong to the ribosome protection gene (Roberts, 1996). Previous studies have shown that efflux pumps play a major role in both organic contaminants tolerance and bioremediation (Fernandes et al., 2003), and combined with KEGG annotation results of specific genes in Pandoraea sp. XY-2, so we selected tetA(48), tetG, and tetR genes for further origin and evolution analysis.
The deviant GC content is used as a detect method of HGT (Xie et al., 2014). The average GC contents of the tetA(48) genes were higher than those of the average of the entire genomes (63.33–65.57 vs. 62.5–64.7) except that the average GC content of the tetA(48) genes in Pandoraea sp. XY-2 was slightly lower than that of the average of the genome (63.72 vs. 63.76) (Figure 9). It is worth noting that the tetG gene and tetR gene were specific genes of Pandoraea sp. XY-2, whose GC content were significantly lower than that of the genome (57.74 vs. 63.76 and 59.17 vs. 63.76, respectively). These data showing variation of GC content between tetracycline resistance genes and the corresponding genomes, which indicated that these tetracycline resistance genes may be acquired in Pandoraea strains by HGT.
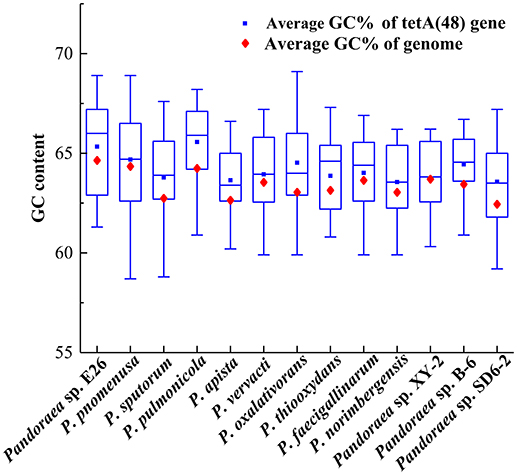
Figure 9. Comparison of GC contents of the tetA(48) genes with those of the average of the entire genomes.
To gain insights into the origin of tetracycline resistance genes in Pandoraea, three NJ phylogenetic trees were constructed based on the tetA(48), tetG, and tetR genes encoding protein sequences (Figures 10–12). As shown in Figure 10, phylogenetic analysis revealed that three tetA(48) genes (XY.2_GM000586, XY.2_GM000605 and XY.2_GM004567) of Pandoraea and tetA(48) genes of Paraburkholderia spp., Burkholderia spp., and Caballeronia spp. were sister groups. It is obviously to note that, the other three tetA(48) genes (XY.2_GM001703, XY.2_GM003133 and XY.2_GM003479) of Pandoraea sp. XY-2 formed monophyletic clade, respectively. Furthermore, the tetG gene from Pandoraea sp. XY-2 grouped to tetG gene of S. enterica, V. cholerae, P. aeruginosa, and Proteobacteria (Figure 11). The tetR gene from Pandoraea sp. XY-2 grouped to tetR gene of S. enterica, V. cholerae, A. baumannii, and F. marinus (Figure 12). These results imply that three tetA(48) genes (XY.2_GM000586, XY.2_GM004567, and XY.2_GM000605) may be acquired via HGT from members of Paraburkholderia, Burkholderia, and Caballeronia. While, the other three tetA(48) genes (XY.2_GM001703, XY.2_GM003133, and XY.2_GM003479) may be acquired via HGT from unidentified sources in early evolutionary history. The tetG gene may be acquired via HGT from S. enterica, V. cholerae, P. aeruginosa and Proteobacteria, and tetR from S. enterica, V. cholerae, A. baumannii, and F. marinus.
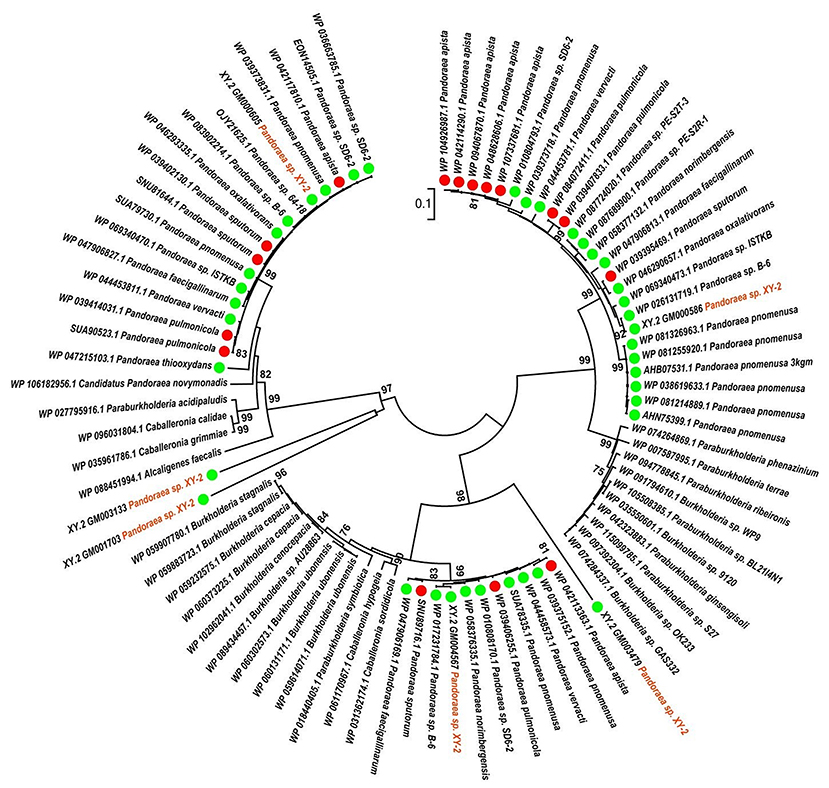
Figure 10. Neighbor joining phylogenetic tree of 6 tetA(48) genes derived from Pandoraea sp. XY-2 and other representative species. Bootstrap values are indicated at each node based on a total of 1,000 bootstrap replicates. The genes of Pandoraea sp. XY-2 were marked in orange. The green dot and red dot indicated that each strain of Pandoraea spp. was isolated from environment and clinical, respectively.
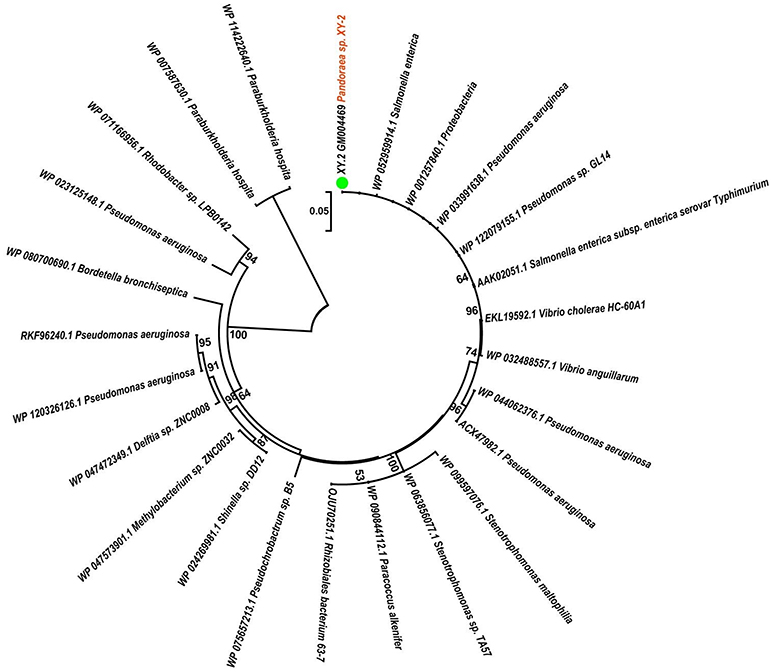
Figure 11. Neighbor joining phylogenetic tree of tetG gene derived from Pandoraea sp. XY-2 and other representative species. Bootstrap values are indicated at each node based on a total of 1,000 bootstrap replicates. The genes of Pandoraea sp. XY-2 were marked in orange. The green dot indicated that Pandoraea sp. XY-2 was isolated from environment.
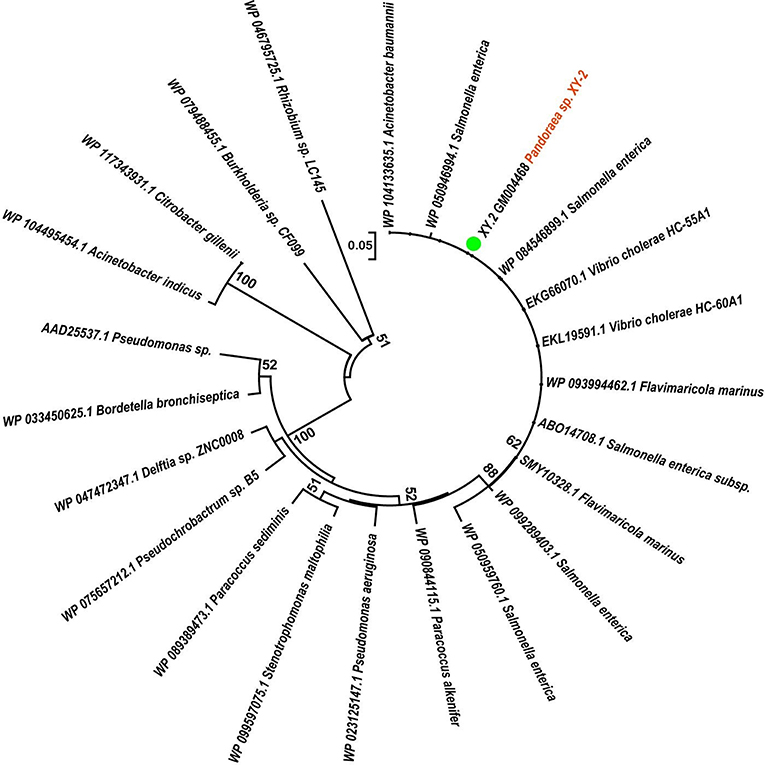
Figure 12. Neighbor joining phylogenetic tree of tetR gene derived from Pandoraea sp. XY-2 and other representative species. Bootstrap values are indicated at each node based on a total of 1,000 bootstrap replicates. The genes of Pandoraea sp. XY-2 were marked in orange. The green dot indicated that Pandoraea sp. XY-2 was isolated from environment.
Discussion
The data we present represents a first glimpse into the evolution and mechanism for tetracycline biodegradation and resistance in genus Pandoraea. Different from sulfonamide compounds, whose degradation by pure bacterial cultures have been reported (Tappe et al., 2013), there are only a few microorganisms that can degrade tetracycline. Previous literature reports that Stenotrophomonas maltophilia strain DT1 can effectively biotransform tetracycline, which can degrade about 46% tetracycline at 30°C and pH 9 in 7 days (Leng et al., 2016). Huang et al. (2016) observed that Trichosporon mycotoxinivorans XPY-10 which was isolated from wastewater could degrade about 46.28% tetracycline at 34°C within 7 days. Zhang et al. (2015) reports that 57.8% tetracycline could be degraded by Advenella kashmirensi at 30°C and pH 7 in 6 days. A recent study showed that a maximum tetracycline degradation ratio of 78.78% can be obtained by Klebsiella sp. SQY5 at an initial pH of 7.17, a temperature of 34.96°C, an initial tetracycline concentration of 61.27 mg L−1 and an inoculation dose of 29.89% (v/v) using SAS software prediction (Shao et al., 2018). In this study, Pandoraea sp. XY-2 was screened out, which could biodegrade 74% tetracycline at pH 7.0 and 30°C within 6 days. At present, the functional characteristics of Pandoraea have been reported. Previously identified Pandoraea spp. are predominantly clinical isolates with a few strains being associated with biodegradation of environmental pollutants (Lim et al., 2016). For example, Pandoraea participates in the bioremediation of lignin, plasticizer dibutyl phthalate, phenol and petroleum hydrocarbon pollutants (Sarkar et al., 2017; Fang et al., 2018; Liu et al., 2018; Yang et al., 2018). In short, it has not yet been reported that tetracycline could be biodegraded by genus Pandoraea. Therefore, this is the first discovery that Pandoraea sp. XY-2 can efficiently biodegrade tetracycline, and its biodegradation percentage was about 74%, which indicates that Pandoraea sp. XY-2 is an excellent microorganism resource for tetracycline wastewater treatment.
Although the cell density of Pandoraea sp. XY-2 decreased during decline period, the active cells could continue to biodegrade tetracycline. Therefore, the biodegradation percentage of tetracycline increased slightly as the cell density of Pandoraea sp. XY-2 decreased (Figure 1). Similar to the findings from a previous study (Loftin et al., 2008), this study demonstrated that high temperatures accelerated tetracycline hydrolysis. For the temperature tested, the biodegradation percentage of tetracycline does not decrease as biomass decreased drastically from 35 to 40°C (Figure 2). According to the collision theory, higher temperatures lead to higher reaction rates by causing more collisions between particles (Thornton, 1964). Moreover, high pH also affected the removal of tetracycline, when the initial pH was 9, the biomass of Pandoraea sp. XY-2 decreased but the tetracycline biodegradation percentage increased (Figure 3). This may be because tetracycline can form invalid lactone isomers easily under alkaline conditions (Hallingsørensen et al., 2003). The pH value of the sampling location in this study was about 6.8, which was not conducive to the natural degradation of tetracycline. The optimum pH for Pandoraea sp. XY-2 biodegrading tetracycline is 7.0, so it may play an important role in the situ remediation of tetracycline-contaminated environment.
A detailed understanding of the molecular mechanisms that xenobiotic biodegradation and drug-resistance in genus Pandoraea is optimally addressed using truly comprehensive information about their genomes. Now, the whole genome of Pandoraea sp. XY-2 was successively sequenced using the Pacific Biosciences RS II in the present study. Unsurprisingly, the Pandoraea sp. XY-2 exhibited similarities in genome size and GC content compared to the other Pandoraea strains (Table 1). Moreover, the phylogenetic analysis confirmed that Pandoraea sp. XY-2 was most closely related to the Pandoraea sp. 64–18 and Pandoraea sp. B-6, implying that Pandoraea sp. XY-2, Pandoraea sp. 64–18, and Pandoraea sp. B-6 have a common ancestor. With the rapid advances in genomics analysis technology, new measurements such as ANI are being developed to evaluate the genomic similarity between bacteria (Richter and Rossellómóra, 2009). Here, the nucleotide comparison of the whole genome sequence of Pandoraea sp. XY-2 with other Pandoraea species revealed an ANI% value < 85%. An additional, the dDDH% values of Pandoraea sp. XY-2 against all reference strains were less 52.2%. Both ANI and dDDH values described herein were considerably below the threshold for species circumscription (Richter and Rossellómóra, 2009). Therefore, we can consider that Pandoraea sp. XY-2 belongs to a new species.
The CAZymes annotation of Pandoraea sp. XY-2 genome revealed two genes (XY.2_GM002965 and XY.2_GM003795) encoding AA6 family members in its genome sequence. All experimentally characterized proteins in AA6 family were 1,4-benzoquinone reductases, which were intracellular enzymes involved in the biodegradation of aromatic compounds (Spain and Gibson, 1991). It suggested that these enzymes might play an important role in the biodegradation of tetracycline. On the other hand, the COG annotation revealed a large number of genes involved in carbohydrate transport and metabolism, secondary metabolites biosynthesis, transport and catabolism and intracellular trafficking, secretion, and vesicular transport. The existence of these genes may be closely related to the ability of Pandoraea sp. XY-2 to biodegrade tetracycline. The GO annotation results indicated that the protein function of Pandoraea sp. XY-2 was mainly focused on cell composition, enzyme catalysis and metabolism. Furthermore, KEGG annotation showed that 9 genes encoding dioxygenase. Dioxygenase can degrade aromatic compounds by dihydroxylation, monohydroxylation and tandem oxidation (Boyd et al., 2001). Nine GSTs genes were also identified in the genome sequence of Pandoraea sp. XY-2. Previous reports showed that tetracycline can be more readily biodegradable by using the detoxifying enzyme GSTs, and the biotoxicity of tetracycline was greatly reduced (Park and Choung, 2007). The comparative analysis of all the 9 GSTs-encoding genes in Pandoraea sp. XY-2 together with GSTs- encoding genes from close-related strains revealed that the GSTs-encoding genes were present in both environmental and clinical isolates and the contents of these genes did not change significantly. It indicated that there may be no major variation in the ability to degrade tetracycline between these isolates irrespective of their origin. Similar conclusion about environmental strains and clinical ones shared similar number of resistance determinants was also reported by Youenou et al. (2015).
As part of the genomic characterization, the pan and core genome of Pandoraea sp. XY-2 were defined as a way to cluster existing genes in all analyzed genomes. Our pan-genome analysis of strains in the genus Pandoraea revealed that Pandoraea sp. XY-2 had 414 specific genes, which could be related to the unique characteristics of Pandoraea sp. XY-2. As expected, the KEGG annotation of the specific genes in Pandoraea sp. XY-2 revealed 2 important genes tetG and tetR related to tetracycline resistance. The most important example is the tetR gene, which encoding multi-drug binding proteins AcrR. According to previous reports, the multi-drug binding proteins AcrR is member of the TetR family of transcriptional repressors that regulate the expression of the multidrug resistant efflux pumps AcrAB (Routh et al., 2009). In short, these results provided some clues on the specific genes involved in tetracycline resistance development and molecular basis of tetracycline biodegradation capacity in the studied strain of Pandoraea sp. XY-2. On the other hand, the model extrapolation revealed that the number of core genes was relatively stable that an extra genome added would not significantly affect its size, which was consistent with the notion that core genes were conserved genes universally present in all strains (Zhang and Sievert, 2014). Besides, comparison of the complete sequence of the Pandoraea sp. XY-2 and P. apista DSM16535T revealed a total of 67,476 non-synonymous SNPs and 581 InDels were detected in the genome of Pandoraea sp. XY-2. The SNPs, InDels and different syntenial relationships identified here might be associated with differences in biodegradation characteristics of organic contaminants and drug resistance of Pandoraea spp., and also contribute to understanding the phylogeny and population genetics of Pandoraea spp.
In order to providing insights into the molecular basis of tetracycline biodegradation by Pandoraea sp. XY-2, we identified the antibiotic resistance genes by CARD analysis. The results showed that Pandoraea sp. XY-2 was resistant to various antibiotics, suggesting multiple resistance mechanisms presented in Pandoraea sp. XY-2. Particularly, we also identified the efflux pump-encoding genes tetA(48) and tetG and the ribosome protection protein-encoding gene tetB(P) and tetT, which was associated with tetracycline resistance. The efflux pump system can pump tetracycline molecules out of a cell and ribosomal protection proteins can cause an allosteric disruption on tetracycline binding sites and release tetracycline from the ribosome (Thaker et al., 2010; Roberts et al., 2011). Furthermore, efflux pumps play a major role in both solvent tolerance and bioremediation (Fernandes et al., 2003). Therefore, the existence of these resistance genes indicated that the Pandoraea sp. XY-2 had the molecular basis of tetracycline resistance, which makes it possible and precondition to biodegrade tetracycline.
Horizontal gene transfer (HGT) mediated by plasmids, transposons, or phages is considered to be the mechanism responsible for the widespread distribution of biodegradative pathways in bacteria (De and Davies, 2000). The deviant G+C content is one of the indicative used to detect HGT (Xie et al., 2014). We implemented a combination of deviant G+C content detection and phylogenetic analysis to discover putative HGT events since it is difficult to identify HGT events via deviant G+C content alone if HGT happened to occur between the organisms with the similar G+C contents (Hirsch et al., 1995). The different gene content and organization of antibiotic resistance genes suggest complicated evolutionary history of these genes, and also suggest different genetic requirements for effective cellular antibiotic detoxification and regulation of antibiotic resistance operons. Our results supposed that the tetA(48), tetG, and tetR genes in Pandoraea spp. may be acquired by HGT from Paraburkholderia spp., Burkholderia spp., Caballeronia spp., S. enterica, V. cholerae, Proteobacteria, P. aeruginosa, A. baumannii, F. marinus, and some unidentified sources in early evolutionary history. Similar conclusion about gram-positive and gram-negative bacteria tetracycline resistance was a trait acquired via HGT was also reported by Mindlin et al. (2006). Previous studies have shown that Burkholderia cepacia, with genetic determinants for efflux pumps that facilitate tetracycline excretion, was the only bacterium that grew on tetracycline-amended R2A plates (Rysz and Alvarez, 2004). Moreover, Todorovic et al.' 2015 have detected the tetA gene and the corresponding tetR gene in the Salmonella Infantis isolated from 28 poultry farms in the Northern part of Serbia. The determination of tetracycline resistance genes in V. cholerae have also been researched by Khany et al. (2016).
Conclusions
Pandoraea sp. XY-2 has good performance for biodegrade tetracycline, which could biodegrade 74% tetracycline at pH 7.0 and 30°C within 6 days. Genomic analyses including ANI and dDDH provided a clear result that the Pandoraea sp. XY-2 belongs to a new species. Additionally, the analysis of genome characteristics and comparative genome revealed that Pandoraea sp. XY-2 contained AA6 family members-encoding genes, GSTs-encoding genes and some tetracycline resistance genes, such as tetA(48), tetB(P), tetT, tetG, and tetR, which were relevant to tetracycline biodegradation and resistance. In the near future, the transcriptomics and proteomic technology will be conducted to further explore the molecular mechanism of tetracycline biodegradation by Pandoraea sp. XY-2.
Data Availability
The sequence data of the whole genome of Pandoraea sp. XY-2 have been deposited to NCBI data base, with the accession number of CP030849.
Author Contributions
XuW, XiW, and WZ designed and coordinated the study and carried out the data analysis. XuW, XiW, LS, and WZ performed the bioinformatics analysis. XuW, XiW, JL, and RY carried out the experiments and interpreted data for the work. XuW, XiW, and WZ wrote the manuscript. GQ checked and edited the manuscript. All authors have read and approved the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31470230, 51320105006, 51604308), the Youth Talent Foundation of Hunan Province of China (No. 2017RS3003), Natural Science Foundation of Hunan Province of China (No. 2018JJ2486), Key Research and Development Projects in Hunan Province (2018WK2012), Fundamental Research Funds for the Central Universities of Central South University (No. 2018zzts767).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00033/full#supplementary-material
References
Alsaari, N., Gao, F., Rohul, A. A., Sato, K., Sato, K., Mino, S., et al. (2015). Advanced microbial taxonomy combined with genome-based-approaches reveals that Vibrio astriarenae sp. nov., an Agarolytic Marine Bacterium, forms a new clade in Vibrionaceae. PLoS ONE 10:e0136279. doi: 10.1371/journal.0136279
Baptista, I. I., Zhou, N. Y., Emanuelsson, E. A., Peeva, L. G., Mantalaris, A., and Livingston, A. G. (2010). Evidence of species succession during chlorobenzene biodegradation. Biotechnol. Bioeng. 99, 68–74. doi: 10.1002/bit.21576
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bound, J. P., and Voulvoulis, N. (2004). Pharmaceuticals in the aquatic environment—a comparison of risk assessment strategies. Chemosphere 56, 1143–1155. doi: 10.1016/j.chemosphere.2004.05.010
Boyd, D. R., Sharma, N. D., and Allen, C. C. (2001). Aromatic dioxygenases: molecular biocatalysis and applications. Curr. Opin. Biotech. 12, 564–573. doi: 10.1016/S0958-1669(01)00264-6
Chaisson, M. J., and Tesler, G. (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics13:238. doi: 10.1186/1471-2105-13-238
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chee-Sanford, J. C., Aminov, R. I., Krapac, I. J., Garrigues-Jeanjean, N., and Mackie, R. I. (2001). Occurrence and diversity of tetracycline resistance genes in lagoons and groundwater underlying two swine production facilities. Appl. Environ. Microbiol. 67, 1494–1502. doi: 10.1128/AEM.67.4.1494-1502.2001
Chen, N. (2004). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4.10.1–4.10.14. doi: 10.1002/0471250953.bi0410s25
Coenye, T., Falsen, E., Hoste, B., Ohlén, M., Goris, J., Govan, J. R., et al. (2000). Description of Pandoraea gen. nov. with Pandoraea apista sp. nov., Pandoraea pulmonicola sp. nov., Pandoraea pnomenusa sp. nov., Pandoraea sputorum sp. nov. and Pandoraea norimbergensis comb. nov. Int. J. Syst. Evol. Microbiol. 50(Pt 2):887. doi: 10.1099/00207713-50-2-887
Daghrir, R., and Drogui, P. (2013). Tetracycline antibiotics in the environment: a review. Environ. Chem. Lett. 11, 209–227. doi: 10.1007/s10311-013-0404-8
Daneshvar, M. I., Hollis, D. G., Steigerwalt, A. G., Whitney, A. M., Spangler, L., Douglas, M. P., et al. (2001). Assignment of CDC weak oxidizer group 2 (WO-2) to the genus Pandoraea and characterization of three new Pandoraea genomospecies. J. Clin. Microbiol. 39, 1819–1826. doi: 10.1128/JCM.39.5.1819-1826.2001
De, L. C. F., and Davies, J. (2000). Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol. 8, 128–133. doi: 10.1016/S0966-842X(00)01703-0
Dhillon, B. K., Laird, M. R., Shay, J. A., Winsor, G. L., Lo, R., Nizam, F., et al. (2015). IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 43, 104–108. doi: 10.1093/nar/gkv401
Dhindwal, S., Patil, D. N., Mohammadi, M., Sylvestre, M., Tomar, S., and Kumar, P. (2011). Biochemical studies and ligand-bound structures of biphenyl dehydrogenase from Pandoraea pnomenusa strain B-356 reveal a basis for broad specificity of the enzyme. J. Biol. Chem. 286, 37011–37022. doi: 10.1074/jbc.M111.291013
Fang, X., Li, Q., Lin, Y., Lin, X., Dai, Y., Guo, Z., et al. (2018). Screening of a microbial consortium for selective degradation of lignin from tree trimmings. Bioresour. Technol. 254, 247–255. doi: 10.1016/j.biortech.2018.01.058
Fernandes, P., Ferreira, B. S., and Cabral, J. M. (2003). Solvent tolerance in bacteria: role of efflux pumps and cross-resistance with antibiotics. Int. J. Antimicrob. Ag. 22, 211–216. doi: 10.1016/S0924-8579(03)00209-7
Ferrarini, M., Moretto, M., Ward, J. A., Šurbanovski, N., Stevanović, V., Giongo, L., et al. (2013). An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome. BMC Genomics 14:670. doi: 10.1186/1471-2164-14-670
Gillings, M. R., and Stokes, H. W. (2012). Are humans increasing bacterial evolvability? Trends Ecol. Evol. 27, 346–352. doi: 10.1016/j.tree.2012.02.006
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Micr. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Hallingsørensen, B., Lykkeberg, A., Ingerslev, F., Blackwell, P., and Tjørnelund, J. (2003). Characterisation of the abiotic degradation pathways of oxytetracyclines in soil interstitial water using LC-MS-MS. Chemosphere 50, 1331–1342. doi: 10.1016/S0045-6535(02)00766-X
Harris, R. S. (2007). Improved pairwise alignment of genomic DNA. Dissertat. Theses Gradworks 20, 251–260. doi: 10.1016/j.midw.2003.12.011
Hirsch, A. M., Mckhann, H. I., Reddy, A., Liao, J., Fang, Y., and Marshall, C. R. (1995). Assessing horizontal transfer of nifHDK genes in eubacteria: nucleotide sequence of nifK from Frankia strain HFPCcI3. Mol. Biol. Evol. 12, 16–27. doi: 10.1093/oxfordjournals.molbev.a040184
Huang, X., Zhang, X., Feng, F., and Xu, X. (2016). Biodegradation of tetracycline by the yeast strain Trichosporon mycotoxinivorans XPY-10. Prep Biochem. Biotechnol. 46, 15–22. doi: 10.1080/10826068.2014.970692
Huang, Y. T., Chen, J. M., Ho, B. C., Wu, Z. Y., Kuo, R. C., and Liu, P. Y. (2018). Genome sequencing and comparative analysis of Stenotrophomonas acidaminiphila reveal evolutionary insights into sulfamethoxazole resistance. Front. Microbiol. 9:1013. doi: 10.3339/fmicb.2018.01013
Huber, W. G. (1971). Antibacterial drugs as environmental contaminants. Adv. Environ. Sci. Technol. 2, 289–320.
Jiao, S., Zheng, S., Yin, D., Wang, L., and Chen, L. (2008). Aqueous photolysis of tetracycline and toxicity of photolytic products to luminescent bacteria. Chemosphere 73, 377–382. doi: 10.1016/j.chemosphere.2008.05.042
Jongsik, C., Jae-Hak, L., Yoonyoung, J., Myungjin, K., Seil, K., Byung Kwon, K., et al. (2007). EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int. J. Syst. Evol. Micr. 57, 2259–2261. doi: 10.1099/ijs.0.64915-0
Jung, J., Choi, S., Chun, J., and Park, W. (2012). Genome sequence of pectin-degrading Alishewanella aestuarii strain B11(T), isolated from tidal flat sediment. J. Bacteriol. 194:5476. doi: 10.1128/JB.01255-12
Khany, A., Salehi, M., and Farzami, M. R. (2016). Determination of tetracycline resistance genes in Vibrio cholerae O1 biotype El Tor serotype Inaba strains isolated from outbreaks occurred in Iran in 2013. Int. J. Med. Res. Health Sci. 5, 121–126.
Kim, H. S., Lee, B. Y., Won, E. J., Han, J., Hwang, D. S., Park, H. G., et al. (2015). Identification of xenobiotic biodegradation and metabolism-related genes in the copepod Tigriopus japonicus whole transcriptome analysis. Mar. Genomics 24, 207–208. doi: 10.1016/j.margen.2015.05.011
Konstantinidis, K. T., and Tiedje, J. M. (2007). Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr. Opin. Microbiol. 10, 504–509. doi: 10.1016/j.mib.2007.08.006
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Leng, Y., Bao, J., Chang, G., Zheng, H., Li, X., Du, J., et al. (2016). Biotransformation of tetracycline by a novel bacterial strain Stenotrophomonas maltophilia DT1. J. Hazard. Mater. 318, 125–133. doi: 10.1016/j.jhazmat.2016.06.053
Leng, Y., Bao, J., Song, D., Li, J., Ye, M., and Li, X. (2017). Background nutrients affect the biotransformation of tetracycline by Stenotrophomonas maltophilia as revealed by genomics and proteomics. Environ. Sci. Technol. 51, 10476–10484. doi: 10.1021/acs.est.7b02579
Leong, S., Li, D., Hapgood, K., Zhang, X., and Wang, H. (2016). Ni(OH)2 decorated rutile TiO2 for efficient removal of tetracycline from wastewater. Appl. Catal. B Environ. 198, 224–233. doi: 10.1016/j.apcatb.2016.05.043
Leski, T. A., Bangura, U., Jimmy, D. H., Ansumana, R., Lizewski, S. E., Stenger, D. A., et al. (2013). Multidrug-resistant tet(X)-containing hospital isolates in Sierra Leone. Int. J. Antimicrob. Ag. 42, 83–86. doi: 10.1016/j.ijantimicag.2013.04.014
Lim, Y. L., Ee, R., Yong, D., Yu, C. Y., Ang, G. Y., Tee, K. K., et al. (2016). Complete genome sequence analysis of Pandoraea pnomenusa type strain DSM 16536T isolated from a cystic fibrosis patient. Front. Microbiol. 7:109. doi: 10.3389/fmicb.2016.00109
Liu, D., Yan, X., Zhuo, S., Si, M., Liu, M., Wang, S., et al. (2018). Pandoraea sp. B-6 assists the deep eutectic solvent pretreatment of rice straw via promoting lignin depolymerization. Bioresour. Technol. 257, 62–68. doi: 10.1016/j.biortech.2018.02.029
Loftin, K. A., Adams, C. D., Meyer, M. T., and Surampalli, R. (2008). Effects of ionic strength, temperature, and pH on degradation of selected antibiotics. J. Environ. Q. 37, 378–386. doi: 10.2134/jeq2007.0230
Mangwani, N., Shukla, S. K., Kumari, S., Rao, T. S., and Das, S. (2015). Characterization of Stenotrophomonas acidaminiphila NCW-702 biofilm for implication in the degradation of polycyclic aromatic hydrocarbons. J. Appl. Microbiol. 117, 1012–1024. doi: 10.1111/jam.12602
Mcarthur, A. G., and Wright, G. D. (2015). Bioinformatics of antimicrobial resistance in the age of molecular epidemiology. Curr. Opin. Microbiol. 27, 45–50. doi: 10.1016/j.mib.2015.07.004
Mesgari Abbasi, M., Babaei, H., Ansarin, M., Nourdadgar, A., and Nemati, M. (2011). Simultaneous determination of tetracyclines residues in bovine milk samples by solid phase extraction and HPLC-FL method. Adv. Pharm. Bull. 1, 34–39. doi: 10.5681/apb.2011.005
Migliore, L., Fiori, M., Spadoni, A., and Galli, E. (2012). Biodegradation of oxytetracycline by Pleurotus ostreatus mycelium: a mycoremediation technique. J. Hazard. Mater. 215–216, 227–232. doi: 10.1016/j.jhazmat.2012.02.056
Mindlin, S. Z., Petrova, M. A., Bass, I. A., and Zhm, G. (2006). Origin, evolution, and migration of drug resistance genes. Russ. J. Genet. 42:1257. doi: 10.1134/S1022795406110081
Nei, M., and Rooney, A. P. (2005). Concerted and birth-and-death evolution of multigene families*. Annu. Rev. Genet. 39, 121–152. doi: 10.1146/annurev.genet.39.073003.112240
Park, H., and Choung, Y. K. (2007). Degradation of antibiotics (Tetracycline, Sulfathiazole, Ampicillin) using enzymes of glutathion S-transferase. Hum. Ecol. Risk Assess. Int. J. 13, 1147–1155. doi: 10.1080/10807030701506223
Parte, A. A. (2014). LPSN–list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 42, 613–616. doi: 10.1093/nar/gkt1111
Rhoads, A., and Au, K. F. (2015). PacBio sequencing and its applications. GenomicsProteomics Bioinformatics 13, 278–289. doi: 10.1016/j.gpb.2015.08.002
Richter, M., and Rossellómóra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Roberts, J. A., Norris, R., Paterson, D. L., and Martin, J. H. (2011). Therapeutic drug monitoring of antimicrobials. Br. J. Clin. Pharm. 73, 27–36. doi: 10.1111/j.1365-2125.2011.04080.x.
Roberts, M. (1996). Tetracycline resistance determinants: mechanisms of action, regulation of expression, genetic mobility, and distribution. FEMS Microbiol. Rev. 19, 1–24. doi: 10.1111/j.1574-6976.1996.tb00251.x
Rosselló-Móra, R., and Amann, R. (2015). Past and future species definitions for bacteria and archaea. Syst. Appl. Microbiol. 38, 209–216. doi: 10.1016/j.syapm.2015.02.001
Routh, M. D., Su, C. C., Zhang, Q., and Yu, E. W. (2009). Structures of AcrR and CmeR: insight into the mechanisms of transcriptional repression and multi-drug recognition in the TetR family of regulators. Biochim. Biophys. Acta 1794, 844–851. doi: 10.1016/j.bbapap.2008.12.001
Rysz, M., and Alvarez, P. J. J. (2004). Amplification and attenuation of tetracycline resistance in soil bacteria: aquifer column experiments. Water Res. 38, 3705–3712. doi: 10.1016/j.watres.2004.06.015
Sarkar, P., Roy, A., Pal, S., Mohapatra, B., Kazy, S. K., Maiti, M. K., et al. (2017). Enrichment and characterization of hydrocarbon-degrading bacteria from petroleum refinery waste as potent bioaugmentation agent for in situ bioremediation. Bioresour. Technol. 242, 15–27. doi: 10.1016/j.biortech.2017.05.010
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686– W689. doi: 10.1093/nar/gki366
Science New York, N. Y. (2000). A whole-genome assembly of Drosophila. Science 287, 2196–2204. doi: 10.1126/science.287.5461.2196
Shao, S., Hu, Y., Cheng, C., Cheng, J., and Chen, Y. (2018). Simultaneous degradation of tetracycline and denitrification by a novel bacterium, Klebsiella sp. SQY5. Chemosphere 209,35–43. doi: 10.1016/j.chemosphere.2018.06.093
Shi, Y., Chai, L., Tang, C., Yang, Z., Zheng, Y., Chen, Y., et al. (2013). Biochemical investigation of kraft lignin degradation by Pandoraea sp. B-6 isolated from bamboo slips. Bioprocess Biosyst. Eng. 36, 1957–1965. doi: 10.1007/s00449-013-0972-9
Spain, J. C., and Gibson, D. T. (1991). Pathway for biodegradation of p-Nitrophenol in a Moraxella sp. Appl. Environ. Microbiol. 57, 812–819.
Speer, B. S., and Salyers, A. A. (1989). Novel aerobic tetracycline resistance gene that chemically modifies tetracycline. J. Bacteriol. 171, 148–153. doi: 10.1128/jb.171.1.148-153.1989
Stackebrandt, E., and Ebers, J. (2006). Taxonomic parameter revisited: tarnished gold standards. Microbiol. Today 33, 152–155.
Stryjewski, M. E., Lipuma, J. J.Jr, M. R., Reller, L. B., and Alexander, B.D. (2003). Sepsis, multiple organ failure, and death due to Pandoraea pnomenusa infection after lung transplantation. J. Clin. Microbiol. 41, 2255–2257. doi: 10.1128/JCM.41.5.2255-2257.2003
Tappe, W., Herbst, M., Hofmann, D., Koeppchen, S., Kummer, S., Thiele, B., et al. (2013). Degradation of sulfadiazine by Microbacterium lacus strain SDZm4, isolated from lysimeters previously manured with slurry from sulfadiazine-medicated pigs. Appl. Environ. Microbiol. 79, 2572–2577. doi: 10.1128/AEM.03636-12
Teng, X., Song, Q., Hu, Y., Song, Z., Ying, J., Li, P., et al. (2016). Whole genomic DNA sequencing and comparative genomic analysis of Arthrospira platensis: high genome plasticity and genetic diversity. DNA Res. 23, 325–338. doi: 10.1093/dnares/dsw023
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Thaker, M., Spanogiannopoulos, P., and Wright, G. D. (2010). The tetracycline resistome. Cell. Mol. Life Sci. 67, 419–431. doi: 10.1007/s00018-009-0172-6
Thornton, E. R. (1964). Rates and equilibria of organc reactions, as treated by statistical, thermodynamis and extrathermodynamic methods. J. Am. Chem. Soc. 86:1273. doi: 10.1021/ja01060a084
Todorovic, D., Velhner, M., Milanov, D., Vidanovic, D., SuvajdzIc, L., Stojanov, I., et al. (2015). Characterization of tetracycline resistance of Salmonella enterica subspecies enterica serovar Infantis isolated from poultry in the northern part of Serbia. Acta Vet. 65, 548–556. doi: 10.1515/acve-2015-0046
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. doi: 10.1016/j.mib.2014.11.016
Wan, Y., Jia, A., Zhu, Z., and Hu, J. (2013). Transformation of tetracycline during chloramination: kinetics, products and pathways. Chemosphere 90, 1427–1434. doi: 10.1016/j.chemosphere.2012.09.001
Wang, L., Wang, J., and Jing, C. (2017). Comparative genomic analysis reveals organization, function and evolution of ars genes in Pantoea spp. Front. Microbiol. 8:471. doi: 10.3389/fmicb.2017.00471
Xie, J. B., Du, Z., Bai, L., Tian, C., Zhang, Y., Xie, J. Y., et al. (2014). Comparative genomic analysis of N2-fixing and non-N2-fixing Paenibacillus spp.: organization, evolution and expression of the nitrogen fixation genes. PLoS Genet. 10:e1004231. doi: 10.1371/journal.pgen.1004231
Yang, J., Guo, C., Liu, S., Liu, W., Wang, H., and Dang, Z. (2018). Characterization of a di-n-butyl phthalate-degrading bacterial consortium and its application in contaminated soil. Environ. Sci. Pollut. Res. 25, 17645–17653. doi: 10.1007/s11356-018-1862-0
Yarza, P., Richter, M., Peplies, J., Euzeby, J., Amann, R., Schleifer, K. H., et al. (2008). The all-species living tree project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst. Appl. Microbiol. 31, 241–250. doi: 10.1016/j.syapm.2008.07.001
Youenou, B., Favrebont,é, S., Bodilis, J., Brothier, E., Dubost, A., Muller, D., et al. (2015). Comparative genomics of environmental and clinical Stenotrophomonas maltophilia strains with different antibiotic resistance profiles. Genome Biol. Evol. 7, 2484–2505. doi: 10.1093/gbe/evv161
Zhang, X. Y., Cai, T. J., and Xu, X. P. (2015). Isolation and identification of a Tetracycline -degrading bacterium and optimizing condition for tetracycline degradation. Biotechnol. Bull. 31, 173–180. doi: 10.13560/j.cnki.biotech.bull.1985.2015.01.026
Zhang, Y., and Sievert, S. M. (2014). Pan-genome analyses identify lineage- and niche-specific markers of evolution and adaptation in Epsilonproteo bacteria. Front. Microbiol. 5:110. doi: 10.3389/fmicb.2014.00110
Keywords: Pandoraea sp. XY-2, tetracycline biodegradation, genome sequencing, comparative genomic, tetracycline resistance
Citation: Wu X, Wu X, Shen L, Li J, Yu R, Liu Y, Qiu G and Zeng W (2019) Whole Genome Sequencing and Comparative Genomics Analyses of Pandoraea sp. XY-2, a New Species Capable of Biodegrade Tetracycline. Front. Microbiol. 10:33. doi: 10.3389/fmicb.2019.00033
Received: 25 October 2018; Accepted: 10 January 2019;
Published: 29 January 2019.
Edited by:
Rakesh Sharma, Institute of Genomics and Integrative Biology (CSIR), IndiaReviewed by:
Vasvi Chaudhry, University of Tubingen, GermanyKelle Cardin Freel, University of Hawaii at Manoa, United States
Copyright © 2019 Wu, Wu, Shen, Li, Yu, Liu, Qiu and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weimin Zeng, zengweimin1024@126.com