Targeting Glioblastoma Stem Cells with 2-Deoxy-D-Glucose (2-DG) Potentiates Radiation-Induced Unfolded Protein Response (UPR)

Abstract
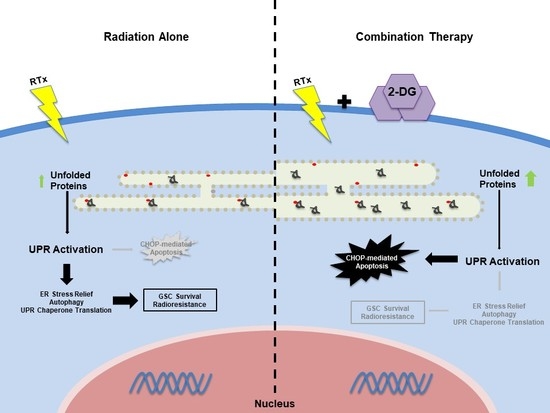
:
1. Introduction
2. Results
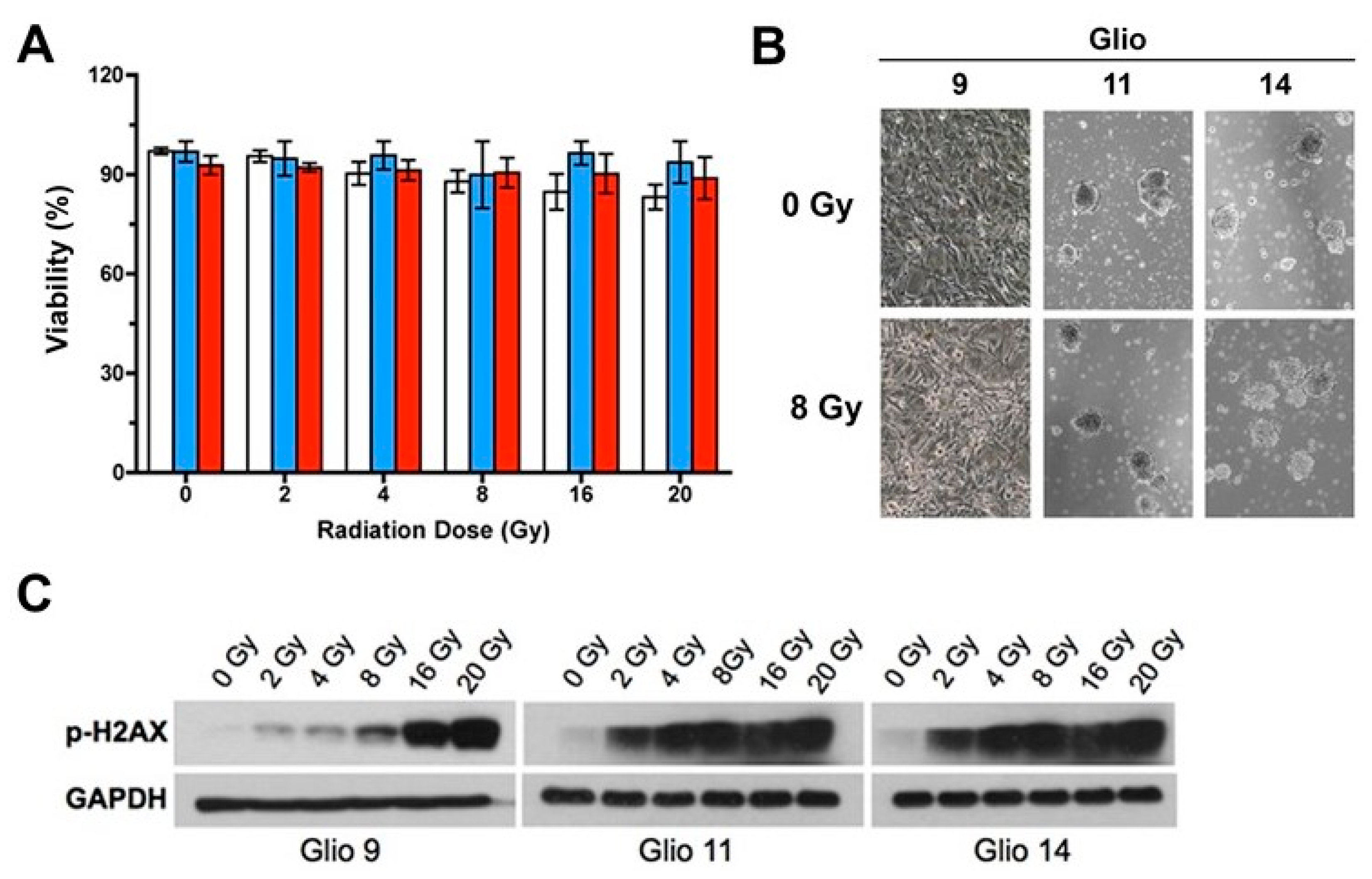
2.1. GSCs Exhibit Robust Radioresistance
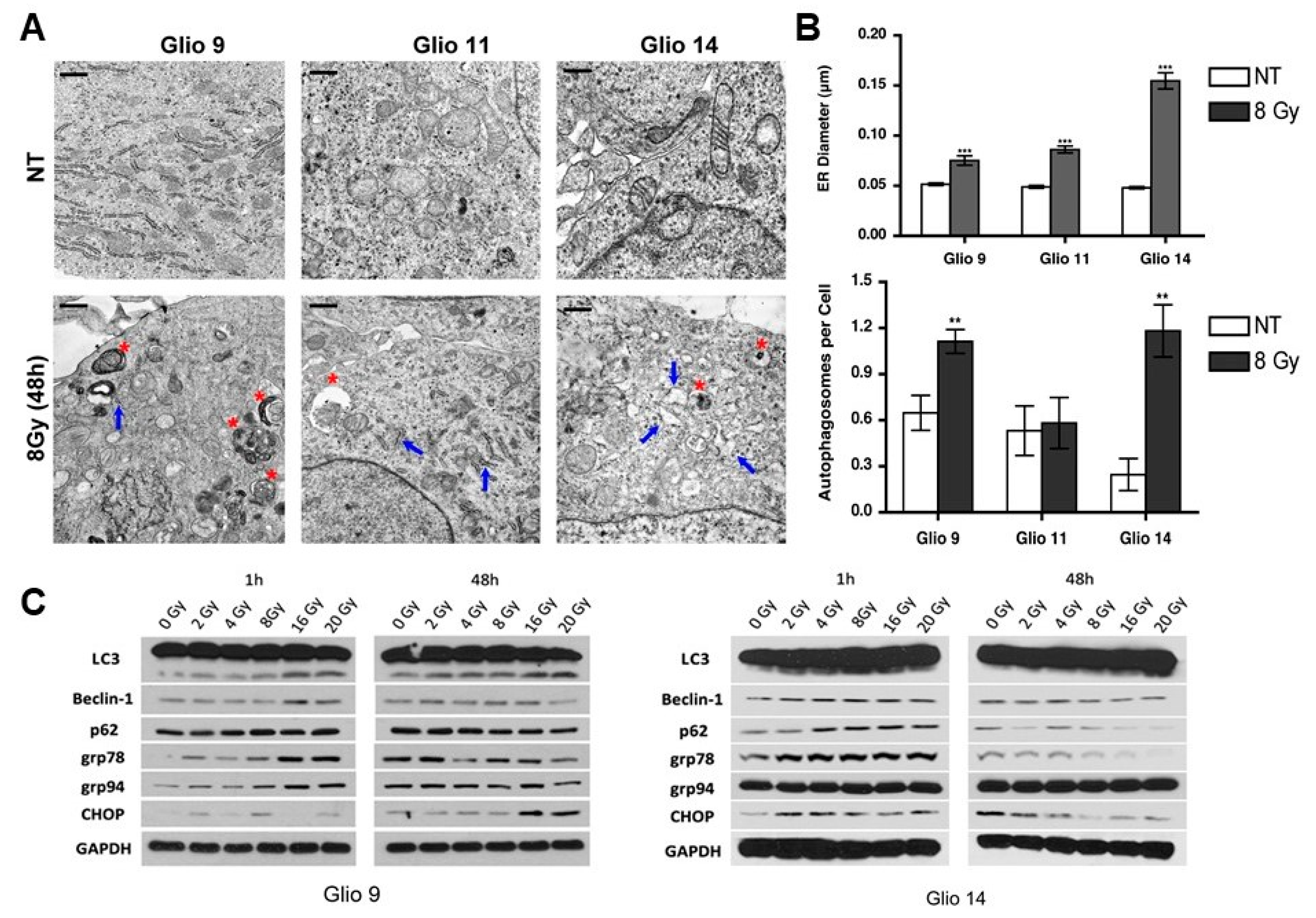
2.2. Radiation Exposure Induces Autophagosome Formation and Activates UPR in GSCs
2.3. Upregulation of UPR Genes in Human GBM Specimen Correlates with Reduced Patient Survival
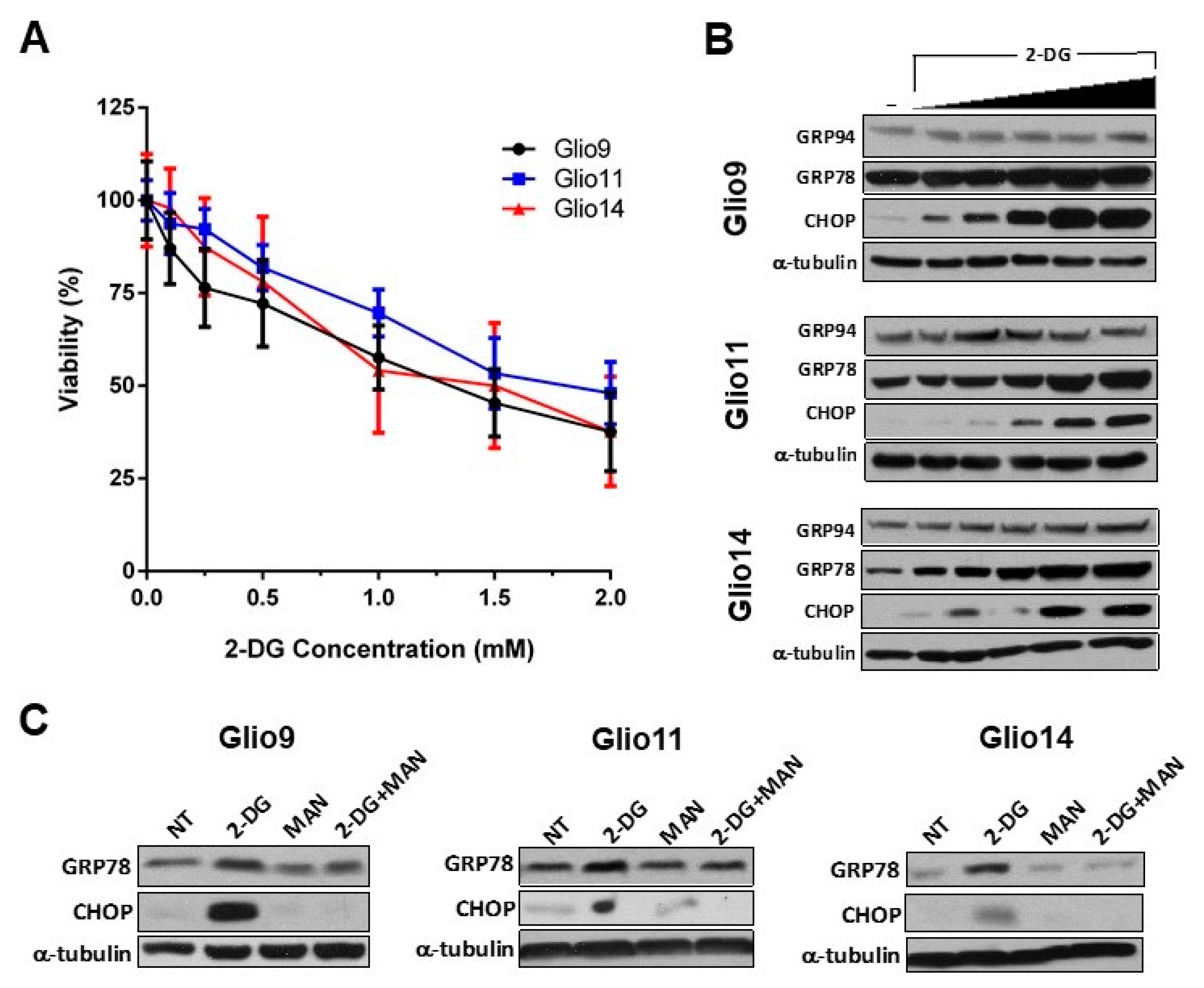
2.4. 2-DG Induces ER Stress in GSCs in a Dose-Dependent Manner
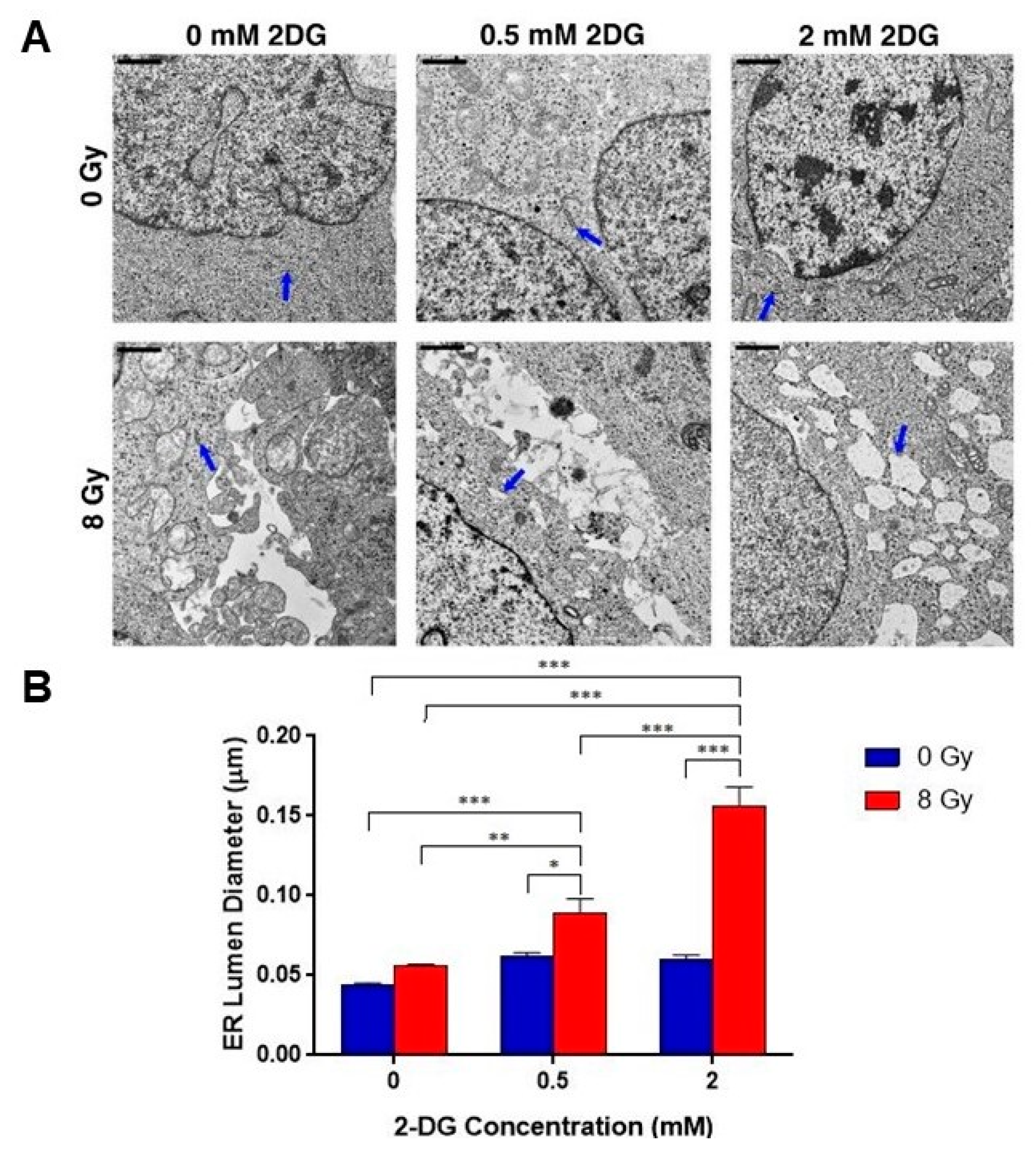
2.5. Combination Radiotherapy and 2-DG Increases ER Dilation in GSCs
2.6. Radiotherapy and 2-DG Cotreatment Potentiates UPR and Leads to Dose-Dependent Loss of Cell Viability and Apoptosis
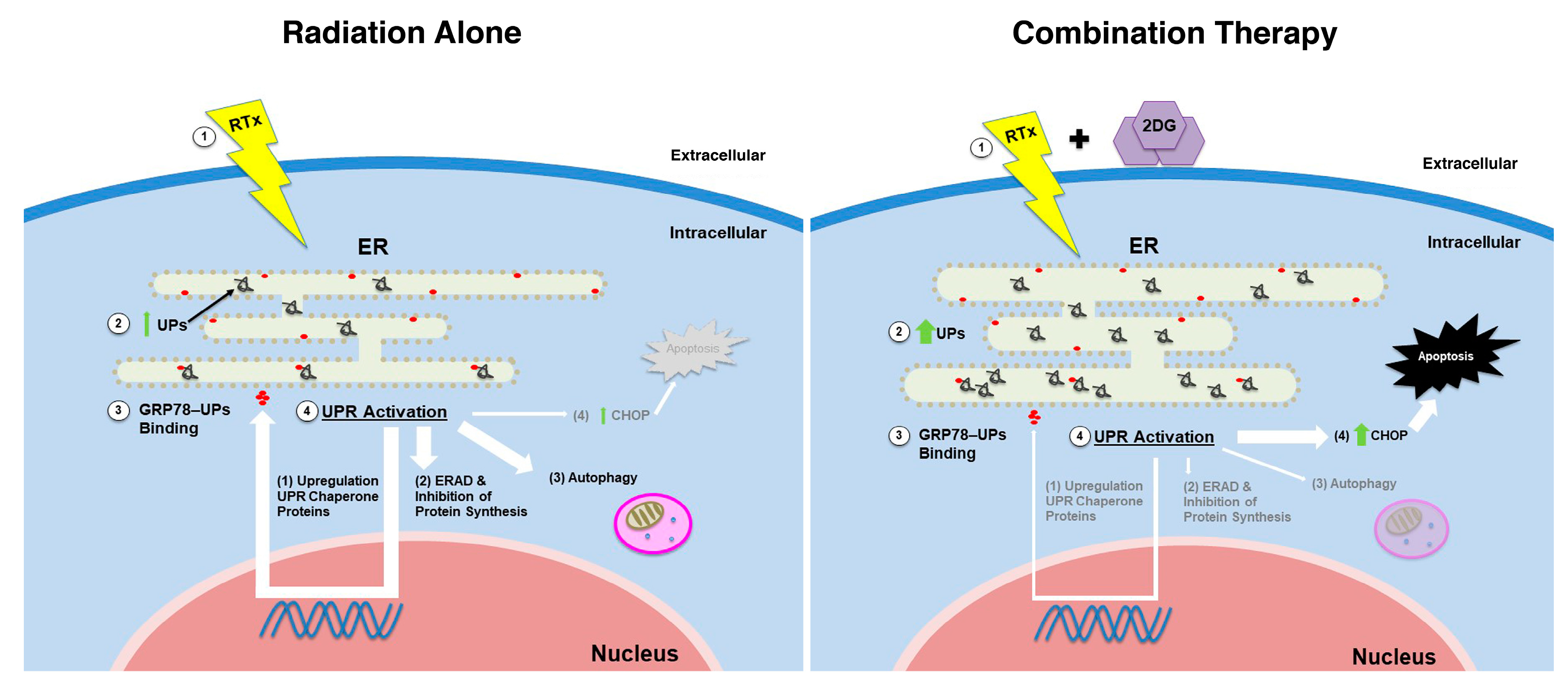
3. Discussion
4. Materials and Methods
4.1. Tissue Culture and Reagents
4.2. Radiation Source
4.3. Viability Assay
4.4. Lactate Dehydrogenase Assay
4.5. Transmission Electron Microscopy (TEM)
4.6. Western Blot
4.7. The Cancer Genome Atlas (TCGA) Data Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Sundar, S.J.; Hsieh, J.K.; Manjila, S.; Lathia, J.D.; Sloan, A. The role of cancer stem cells in glioblastoma. Neurosurg. Focus 2014, 37, E6. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Mbenukui, F.; Johann, D.J. Cancer and the tumor microenvironment: A review of an essential relationship. Cancer Chemother. Pharmacol. 2009, 63, 571–582. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J.; Scheuner, D.; Schroder, M.; Shen, X.; Lee, K.; Liu, C.Y.; Arnold, S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 2002, 3, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Amaravadi, R.K. Targeting the unfolded protein response in cancer. Pharmacol. Res. 2017, 120, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Epple, L.M.; Dodd, R.D.; Merz, A.L.; Dechkovskaia, A.M.; Herring, M.; Winston, B.M.; Lencioni, A.M.; Russell, R.L.; Madsen, H.; Nega, M.; et al. Induction of the unfolded protein response drives enhanced metabolism and chemoresistance in glioma cells. PLoS ONE 2013, 8, e73267. [Google Scholar] [CrossRef] [PubMed]
- Lui, C.Y.; Kaufman, R.J. The unfolded protein response. J. Cell Sci. 2003, 116, 1861–1862. [Google Scholar] [Green Version]
- Zhang, X.Y.; Zhang, T.T.; Song, D.D.; Zhou, J.; Han, R.; Qin, Z.H.; Sheng, R. Endoplasmic reticulum chaperone GRP78 is involved in autophagy activation induced by ischemic preconditioning in neural cells. Mol. Brain 2015, 8, e20. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Glucose-regulated protein in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Lee, A.S. The critical roles in endoplasmic reticulum chaperone and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Act 2012, 1823, 774–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Han, C.; Jiang, Y.; Han, N.; Zhang, M.; Li, G.; Qiao, Q. Inhibition of GRP78 abrogates radioresistance in oropharyngeal carcinoma cells after EGFR inhibition by cetuximab. PLoS ONE 2017, 12, e0188932. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ni, M.; Li, J.; Xiong, S.; Ye, W.; Virrey, J.J.; Mao, C.; Ye, R.; Wang, M.; Pen, L.; et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008, 68, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.; Koh, J.; Kim, S.; Go, H.; Min, H.S.; Kim, Y.A.; Kim, D.K.; Jeon, Y.K.; Chung, D.H. Overexpression of endoplasmic reticulum stress-related proteins, XBP1s and GRP78, predicts poor prognosis in pulmonary adenocarcinoma. Lung Cancer 2018, 122, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Chen, Y.; Huang, S.; Cui, F.; Wang, X. Suppression of GRP78 sensitizes human colorectal cancer cells to oxaliplatin by downregulation of CD24. Oncol. Lett. 2018, 15, 9861–9867. [Google Scholar] [CrossRef] [PubMed]
- Dadey, D.Y.; Kapoor, V.; Khudanyan, A.; Urano, F.; Kim, A.H.; Thotala, D.; Hallahan, D.E. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget 2016, 7, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Dadey, D.Y.A.; Kapoor, V.; Hoye, K.; Khudanyan, A.; Collins, A.; Thotala, D.; Hallahan, D.E. Antibody targeting GRP78 enhances the efficacy of radiation therapy in human glioblastoma and non-small cell lung cancer cell lines and tumor models. Clin. Cancer Res. 2017, 23, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Gerelchuluun, A.; Hong, Z.; Sun, L.; Zenkoh, J.; Moritake, T.; Tsuboi, K. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro Oncol. 2013, 15, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Oslowsli, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar]
- Wang, J.; Takeuchi, T.; Tanaka, S.; Kubo, S.K.; Kayo, T.; Lu, D.; Takata, K.; Koizumi, A.; Izumi, T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J. Clin. Investig. 1999, 103, 27–37. [Google Scholar] [CrossRef]
- Niu, Z.; Wang, M.; Zhou, L.; Yao, L.; Liao, Q.; Zhao, Y. Elevated GRP78 expression is associated with poor prognosis in patients with pancreatic cancer. Sci. Rep. 2015, 5, 16067. [Google Scholar] [CrossRef] [Green Version]
- Nami, B.; Ghasemi-Dizgah, A.; Vaseghi, A. Overexpression of molecular chaperons GRP78 and GRP94 in CD44(hi)/CD24(lo) breast cancer stem cells. Bioimpacts 2016, 6, 105–110. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, Y.; Jia, Z.; Li, Q.; Gong, W.; Wang, L.; Wei, D.; Yao, J.; Fang, S.; Xie, K. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin. Exp. Metastasis 2006, 23, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Mert, I.; Chhina, J.; Allo, G.; Dai, J.; Seward, S.; Carey, M.S.; Llaurado, M.; Giri, S.; Rattan, R.; Munkarah, A.R. Synergistic effect of MEK inhibitor and metformin combination in low grade serous ovarian cancer. Gynecol. Oncol. 2017, 146, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Lee, J.H.; Oh, Y.; Koh, I.; Shim, J.K.; Park, J.; Choi, J.; Yun, M.; Jeon, J.Y.; Huh, Y.M.; et al. Inhibition of glioblastoma tumorspheres by combined treatment with 2-deoxyglucose and metformin. Neuro Oncol. 2017, 19, 197–207. [Google Scholar] [CrossRef]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Barredo, J.C.; Merchan, J.R.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. Endoplasmic reticulum stress induced by 2-deoxyglucose but not glucose starvation activates AMPK through CaMKKβ leading to autophagy. Biochem. Pharmacol. 2013, 85, 1463–1477. [Google Scholar] [CrossRef] [PubMed]
- Kovács, K.; Decatur, C.; Toro, M.; Pham, D.G.; Liu, H.; Jing, Y.; Murray, T.G.; Lampidis, T.J.; Merchan, J.R. 2-deoxy-glucose downregulates endothelial AKT and ARK via interverence with N-linked glycosylation, induction of endoplasmic reticulum stress and GSK3β activation. Mol. Cancer Ther. 2016, 15, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, D.G.; Ledur, P.F.; Abujamra, A.L.; Brunetto, A.L.; Schwartsmann, G.; Lenz, G.; Roesler, R. Cancer stem cells and the biology of brain tumors. Curr. Stem Cell Res. Ther. 2009, 4, 306–313. [Google Scholar] [CrossRef]
- Garg, A.D.; Maes, H.; van Vliet, A.R.; Agostinis, P. Targeting the hallmarks of cancer with therapy-induced endoplasmic reticulum (ER) stress. Mol. Cell. Oncol. 2014, 2, e975089. [Google Scholar] [CrossRef]
- Verfaille, T.; Garg, A.D.; Agostinis, P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013, 332, 249–264. [Google Scholar] [CrossRef]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadey, D.Y.A.; Kapoor, V.; Khudanyan, A.; Thotala, D.; Hallahan, D.E. PERK regulates glioblastoma sensitivity to ER stress although promoting radiation resistance. Mol. Cancer Res. 2018, 16, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.L.; He, G.; Liu, Y.B.; Wang, Y.; Zhang, B. Endoplasmic reticulum stress sensitizes human esophageal cancer cell to radiation. World J. Gastroenterol. 2013, 19, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Yasui, H.; Takeuchi, R.; Nagane, M.; Meike, S.; Nakamura, Y.; Yamamori, T.; Ikenaka, Y.; Kon, Y.; Murotani, H.; Oishi, M.; et al. Radiosensitization of tumor cells through endoplasmic reticulum stress induced by PEGylated nanogel containing cold nanoparticles. Cancer Lett. 2014, 347, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Yang, Z.; Zhang, L.; Wang, Y.; Gong, W.; Liu, Y. Quercetin suppresses DNA double-strand break repair and enhances the radiosensitivity of human ovarian cancer cells via p53-dependent endoplasmic reticulum stress pathway. OncoTargets Ther. 2017, 11, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Dwarakanath, B.S.; Singh, D.; Banerji, A.K.; Sarin, R.; Venkataramana, N.K.; Jalali, R.; Vishwanath, P.N.; Mohanti, B.K.; Tripathi, R.P.; Kalia, V.K.; et al. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: Present status and future prospects. J. Cancer Res. Ther. 2009, 5 (Suppl. 1), S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Banerji, A.K.; Dwarakanath, B.S.; Tripathi, R.P.; Gupta, J.P.; Mathew, T.L.; Ravindranath, T.; Jain, V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther. Onkol. 2005, 181, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Pastori, C.; Daniel, M.; Penas, C.; Volmar, C.H.; Johnstone, A.L.; Brothers, S.P.; Graham, R.M.; Allen, B.; Sarkaria, J.N.; Komotar, R.J.; et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics 2014, 9, 611–620. [Google Scholar] [CrossRef]
- Gersey, Z.C.; Rodriguez, G.A.; Barbarite, E.; Sanchez, A.; Walters, W.M.; Ohaeto, K.C.; Komotar, R.J.; Graham, R.M. Curcumin decreases malignant characteristics of glioblastoma stem cells via induction of reactive oxygen species. BMC Cancer 2017, 17, 99. [Google Scholar] [CrossRef]
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. Monitoring autophagy by electron microscopy in mammalian cells. Methods Enzymol. 2009, 452, 143–164. [Google Scholar] [PubMed]
- Graham, R.M.; Hernandez, F.; Puerta, N.; De Angulo, G.; Webster, K.A.; Vanni, S. Resveratrol augments ER stress and the cytotoxic effects of glycolytic inhibition in neuroblastoma by downregulating Akt in a mechanism independent of SIRT1. Exp. Mol. Med. 2016, 48, e210. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | NT | Rad (8 Gy) | p-Value |
|---|---|---|---|
| ER Diameter (µm) | |||
| Glio9 | 0.055 ± 0.002 µm | 0.075 ± 0.005 µm | p < 0.001 |
| Glio11 | 0.049 ± 0.002 µm | 0.086 ± 0.003 µm | p < 0.0001 |
| Glio14 | 0.048 ± 0.001 µm | 0.154 ± 0.008 µm | p < 0.0001 |
| AV per Cell | |||
| Glio9 | 0.65 ± 0.11 | 1.11 ± 0.08 | p < 0.01 |
| Glio11 | 0.53 ± 0.16 | 0.58 ± 0.17 | ns |
| Glio14 | 0.25 ± 0.10 | 1.18 ± 0.17 | p <0.01 |
| 2-DG Dose | NT | Rad (8 Gy) |
|---|---|---|
| 0.0 mM | 0.045 ± 0.008 µm | 0.056 ± 0.001 µm |
| 0.5 mM | 0.062 ± 0.0023 µm | 0.089 ± 0.01 µm |
| 2.0 mM | 0.060 ± 0.0026 µm | 0.157 ± 0.012 µm |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, S.S.; Rodriguez, G.A.; Musick, A.; Walters, W.M.; de Cordoba, N.; Barbarite, E.; Marlow, M.M.; Marples, B.; Prince, J.S.; Komotar, R.J.; et al. Targeting Glioblastoma Stem Cells with 2-Deoxy-D-Glucose (2-DG) Potentiates Radiation-Induced Unfolded Protein Response (UPR). Cancers 2019, 11, 159. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11020159
Shah SS, Rodriguez GA, Musick A, Walters WM, de Cordoba N, Barbarite E, Marlow MM, Marples B, Prince JS, Komotar RJ, et al. Targeting Glioblastoma Stem Cells with 2-Deoxy-D-Glucose (2-DG) Potentiates Radiation-Induced Unfolded Protein Response (UPR). Cancers. 2019; 11(2):159. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11020159
Chicago/Turabian StyleShah, Sumedh S., Gregor A. Rodriguez, Alexis Musick, Winston M. Walters, Nicolas de Cordoba, Eric Barbarite, Megan M. Marlow, Brian Marples, Jeffrey S. Prince, Ricardo J. Komotar, and et al. 2019. "Targeting Glioblastoma Stem Cells with 2-Deoxy-D-Glucose (2-DG) Potentiates Radiation-Induced Unfolded Protein Response (UPR)" Cancers 11, no. 2: 159. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11020159