Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca2+ Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells

 , , , ,
, , , ,  ,
,
Abstract
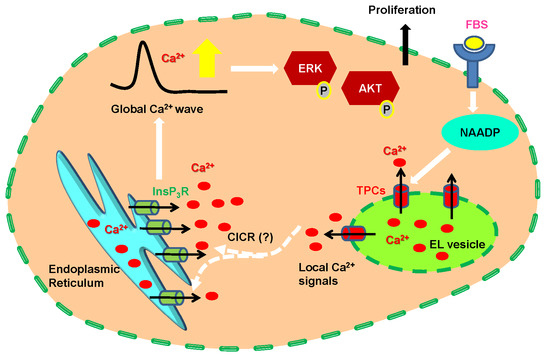
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
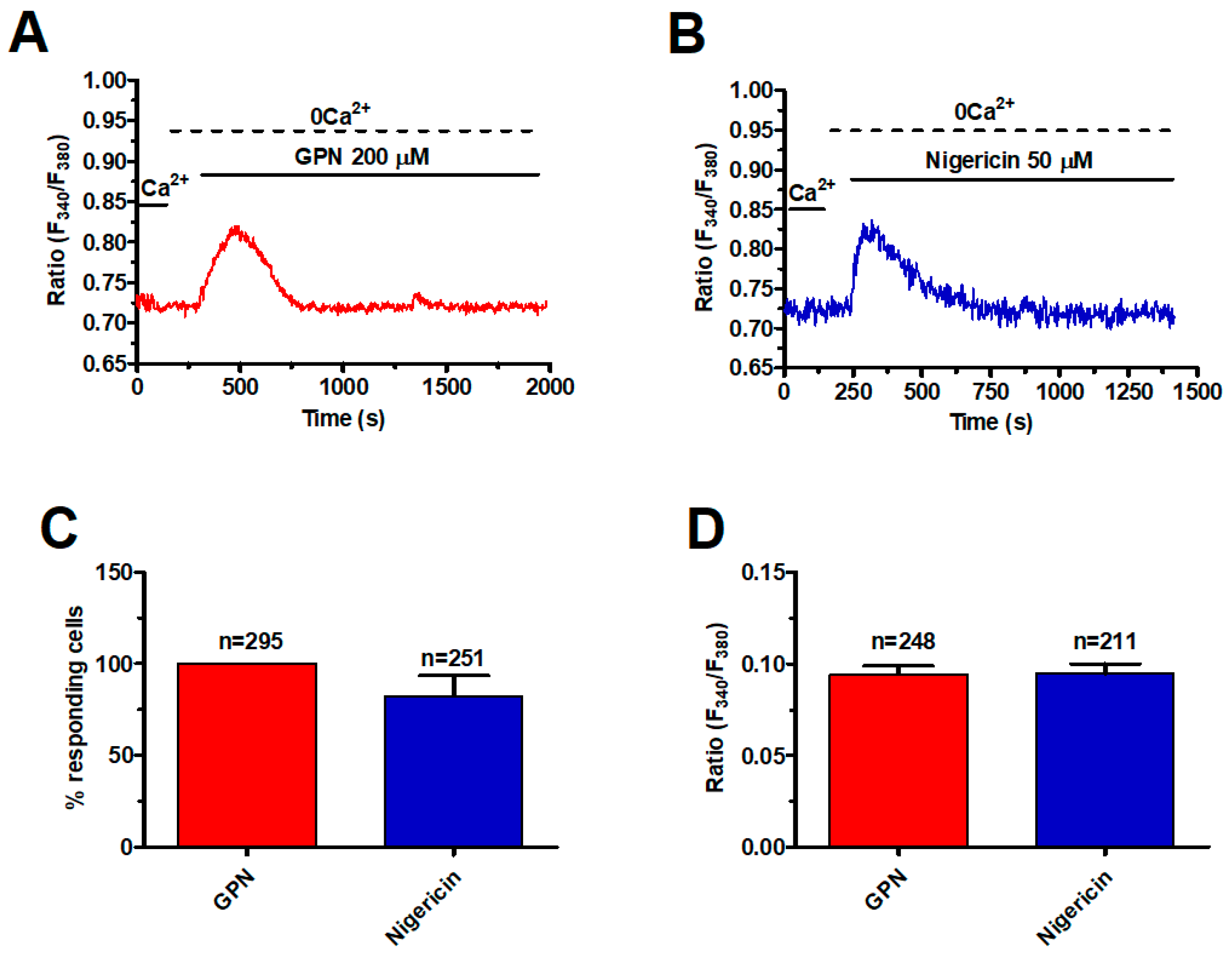
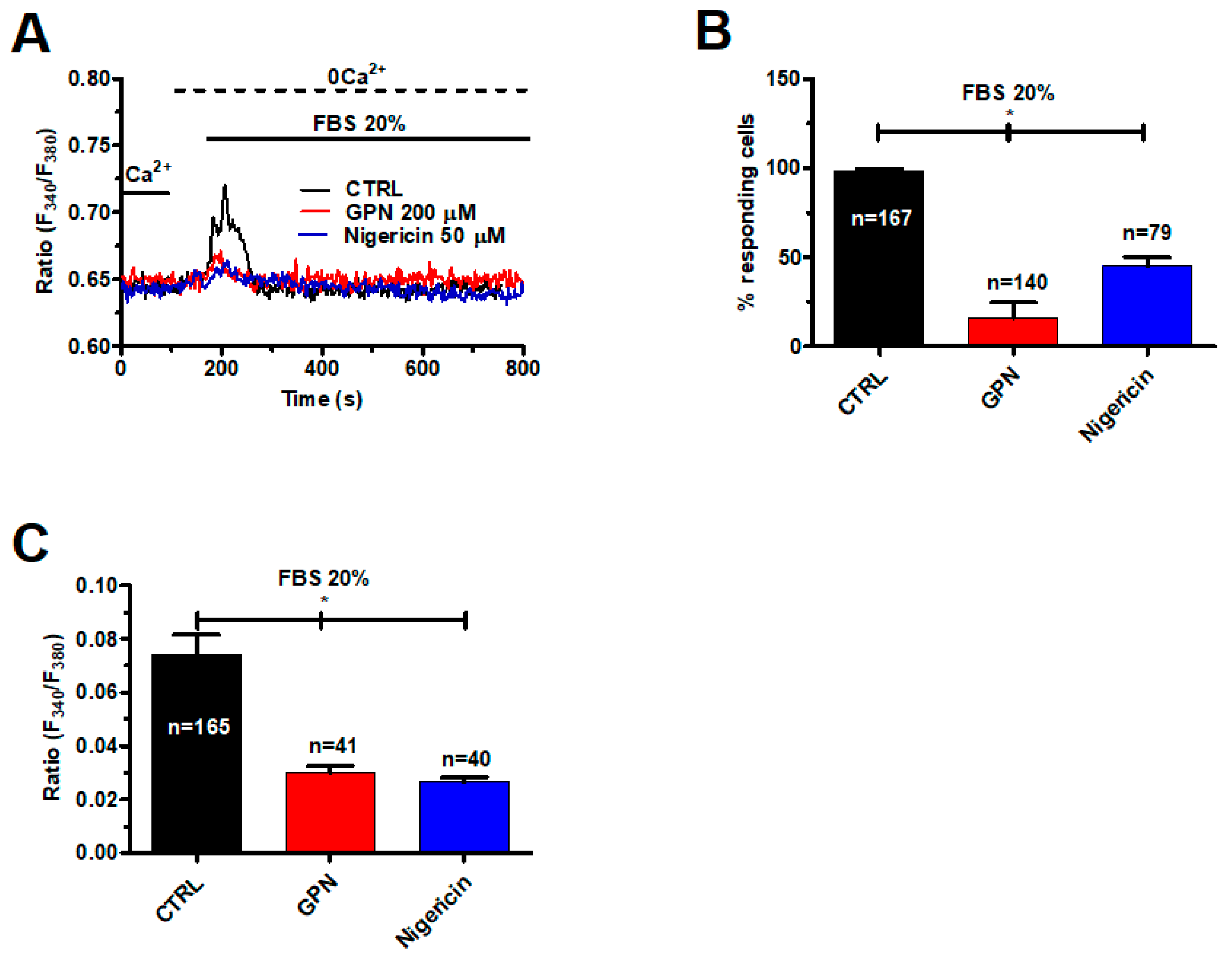
2.1. A Functional Lysosomal Ca2+ Store Is Present in Metastatic Colorectal Cancer Cells
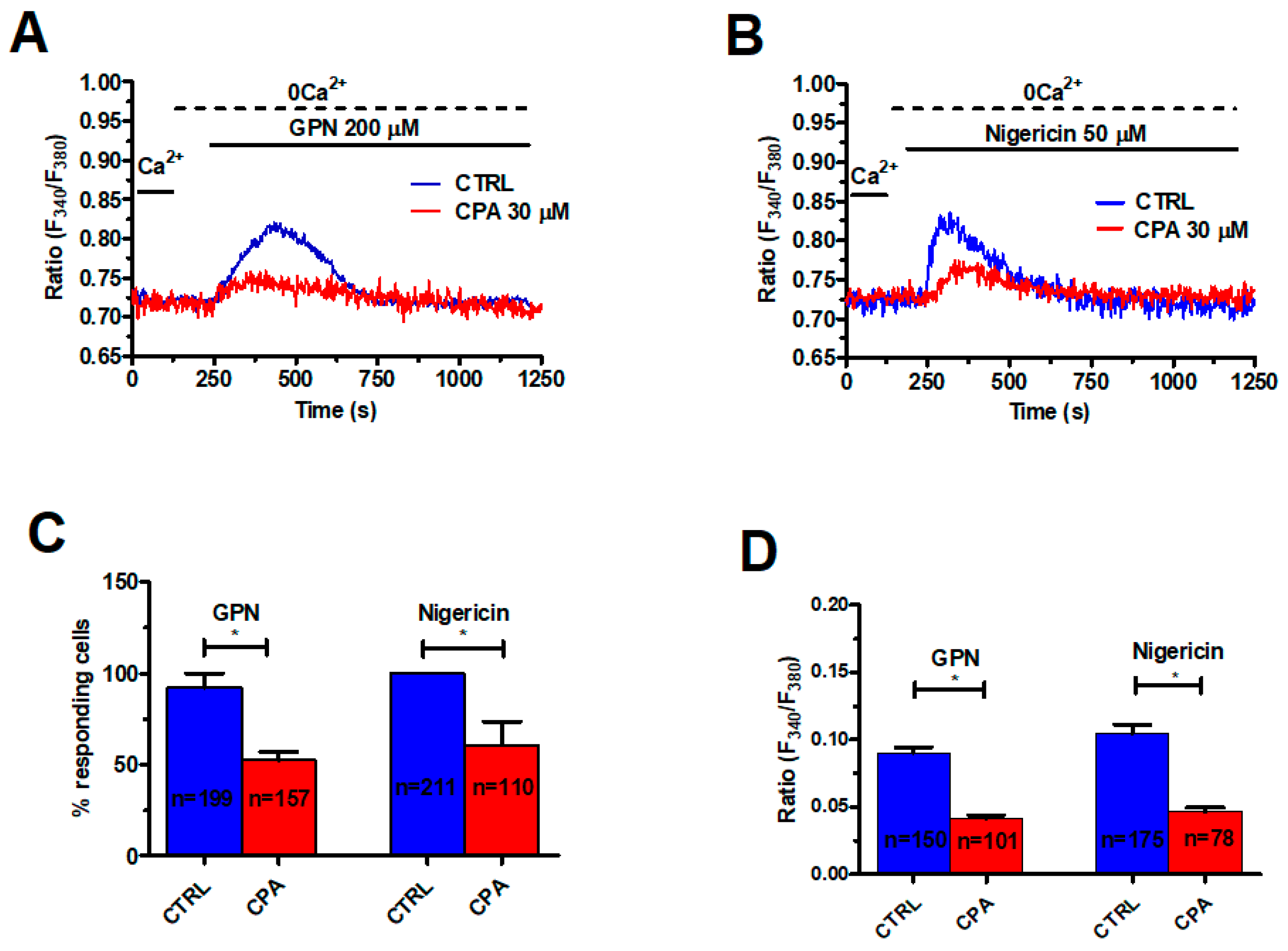
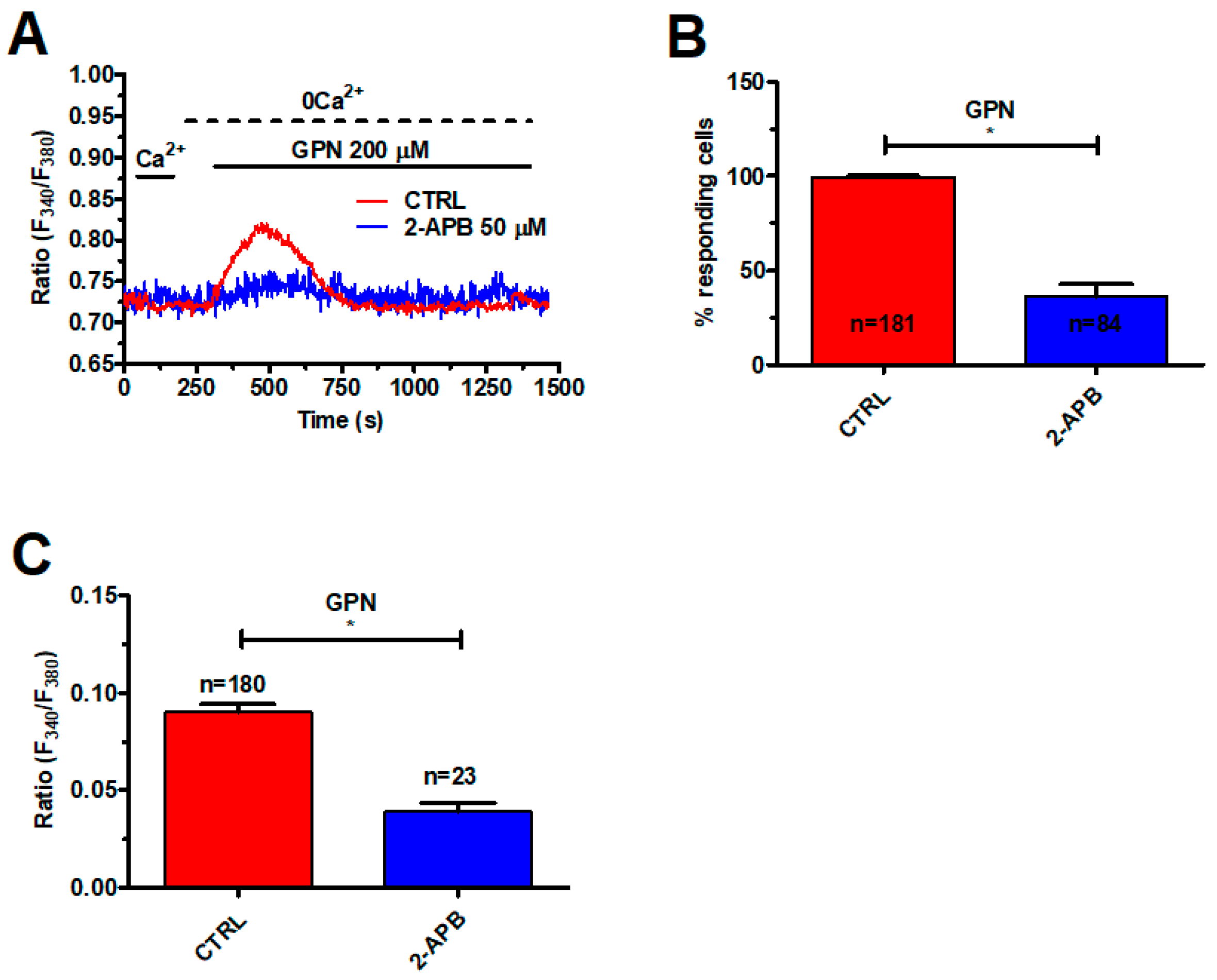
2.2. InsP3-Induced ER Ca2+ Release Sustains the Ca2+ Response to GPN in Metastatic Colorectal Cancer Cells
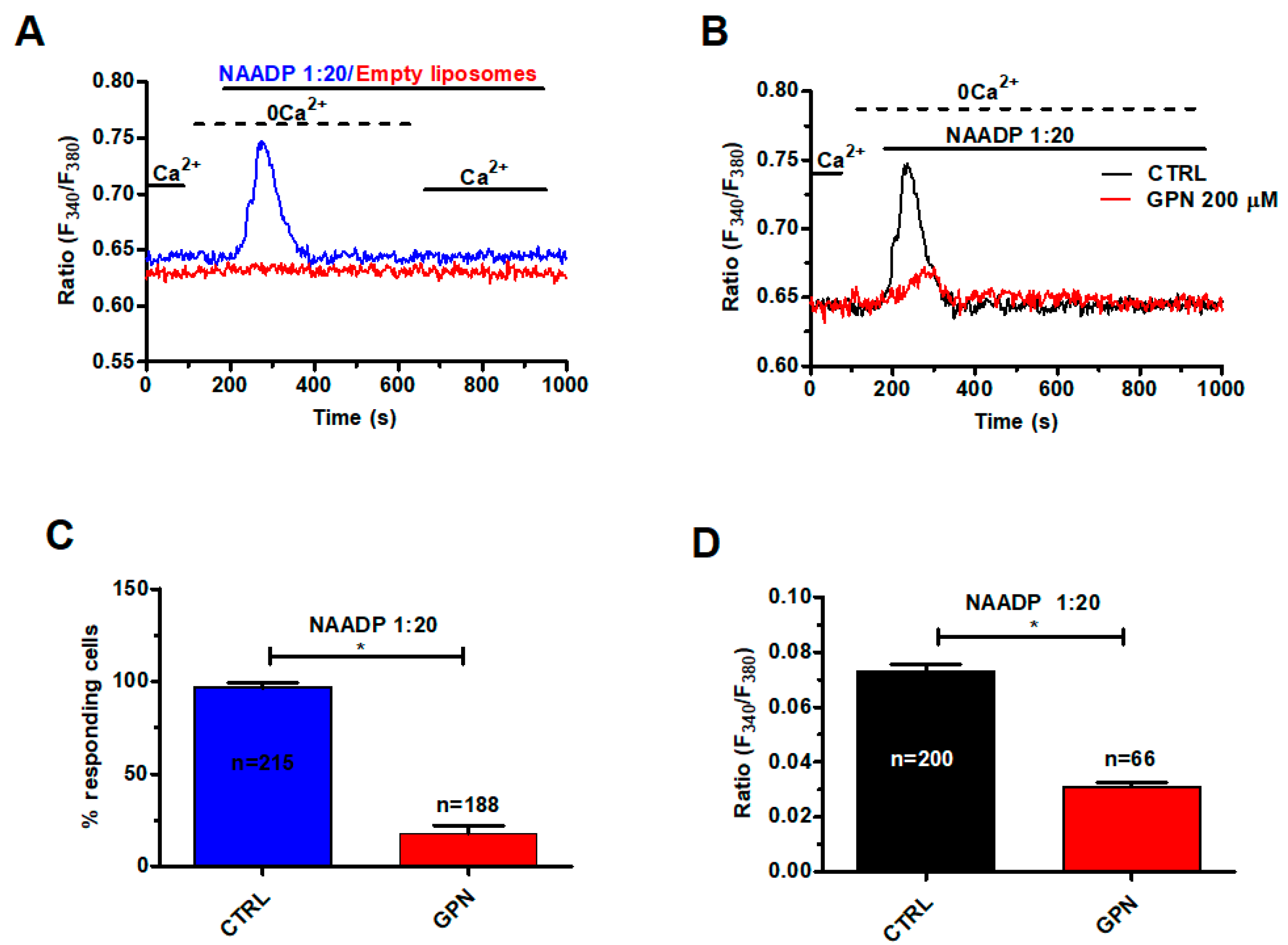
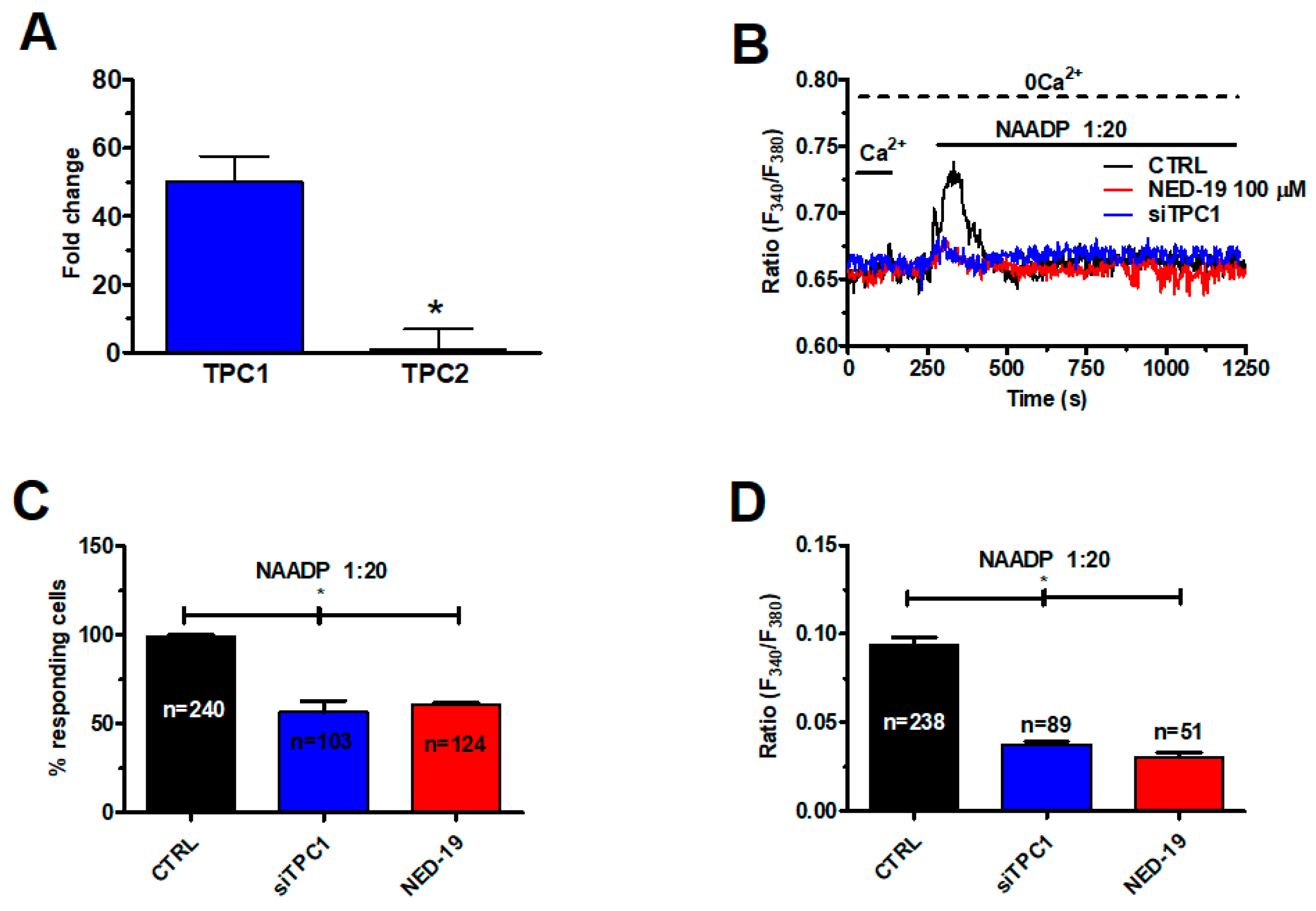
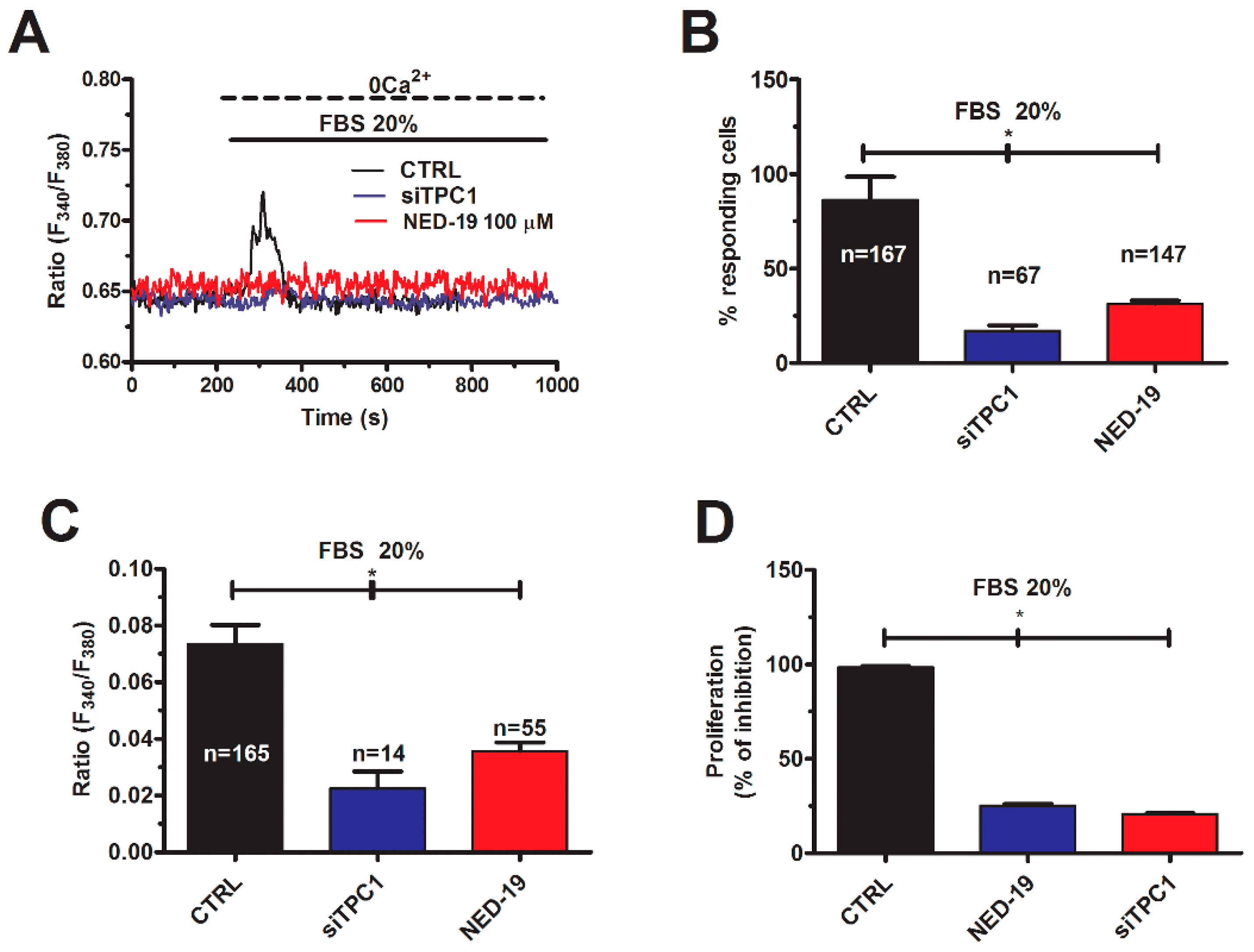
2.3. NAADP Gates TPC1 to Induce Lysosomal Ca2+ Release in Metastatic Colorectal Cancer Cells
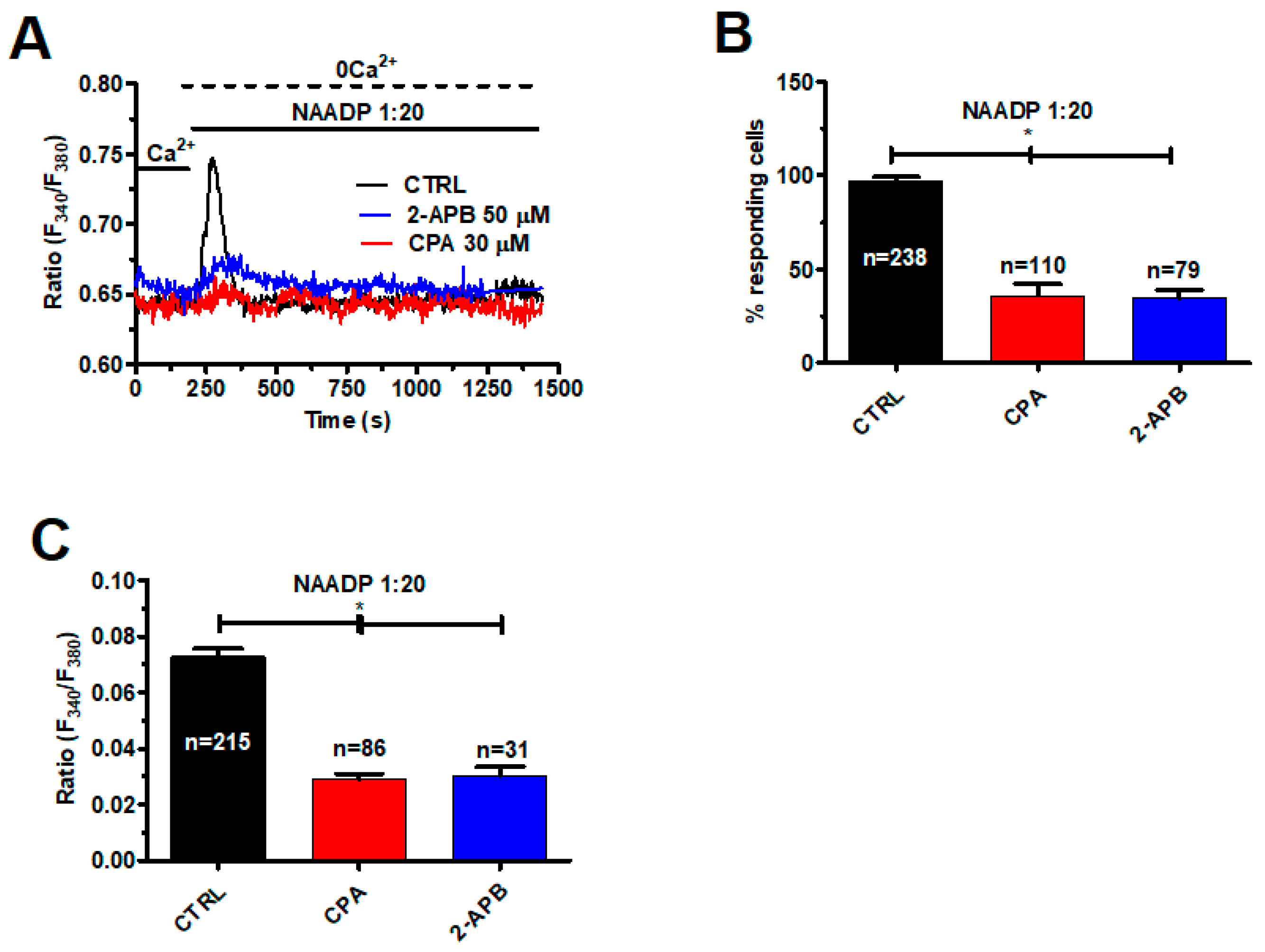
2.4. InsP3-Induced ER Ca2+ Release Sustains NAADP-Induced Intracellular Ca2+ Signals in Metastatic Colorectal Cancer Cells
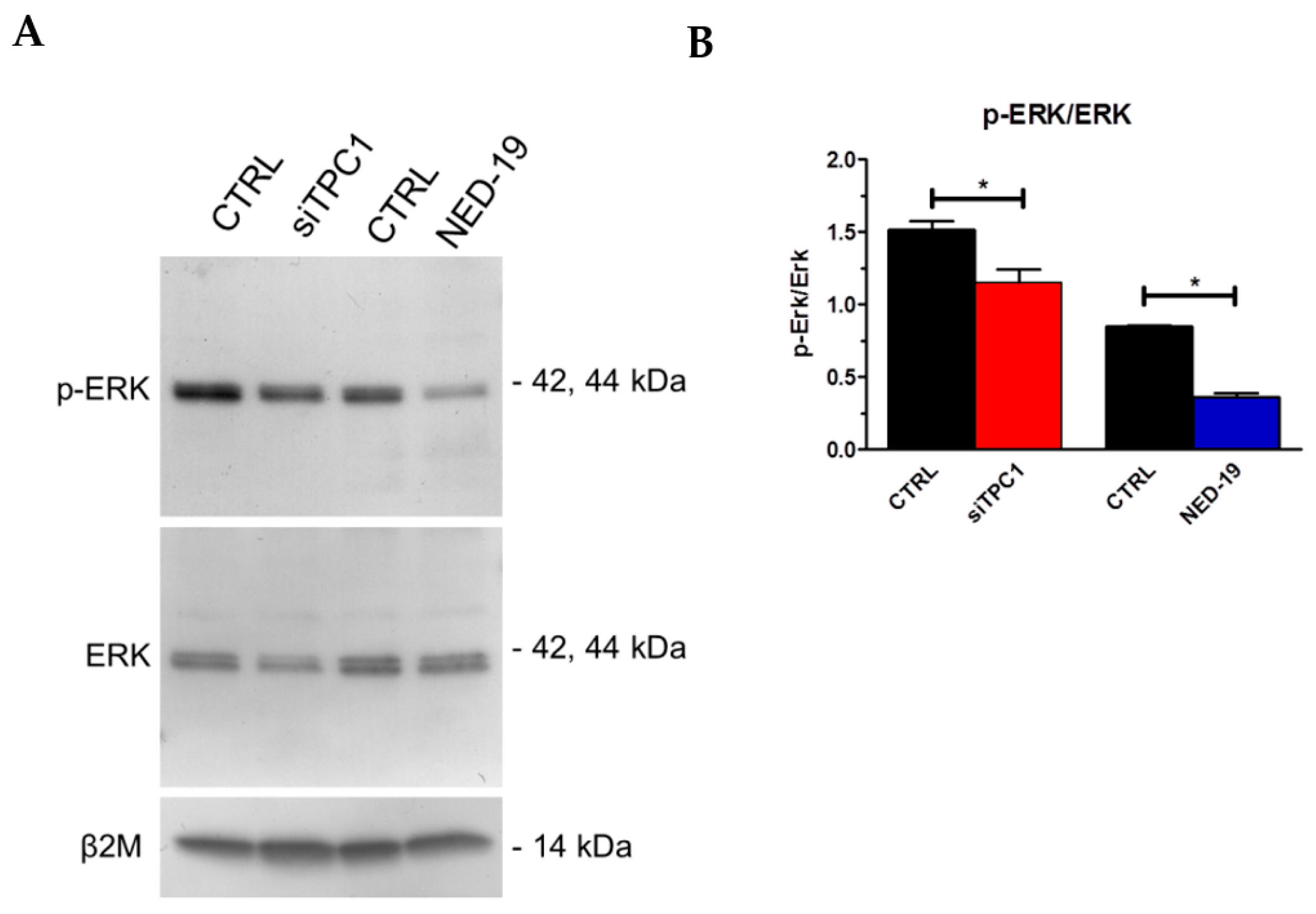
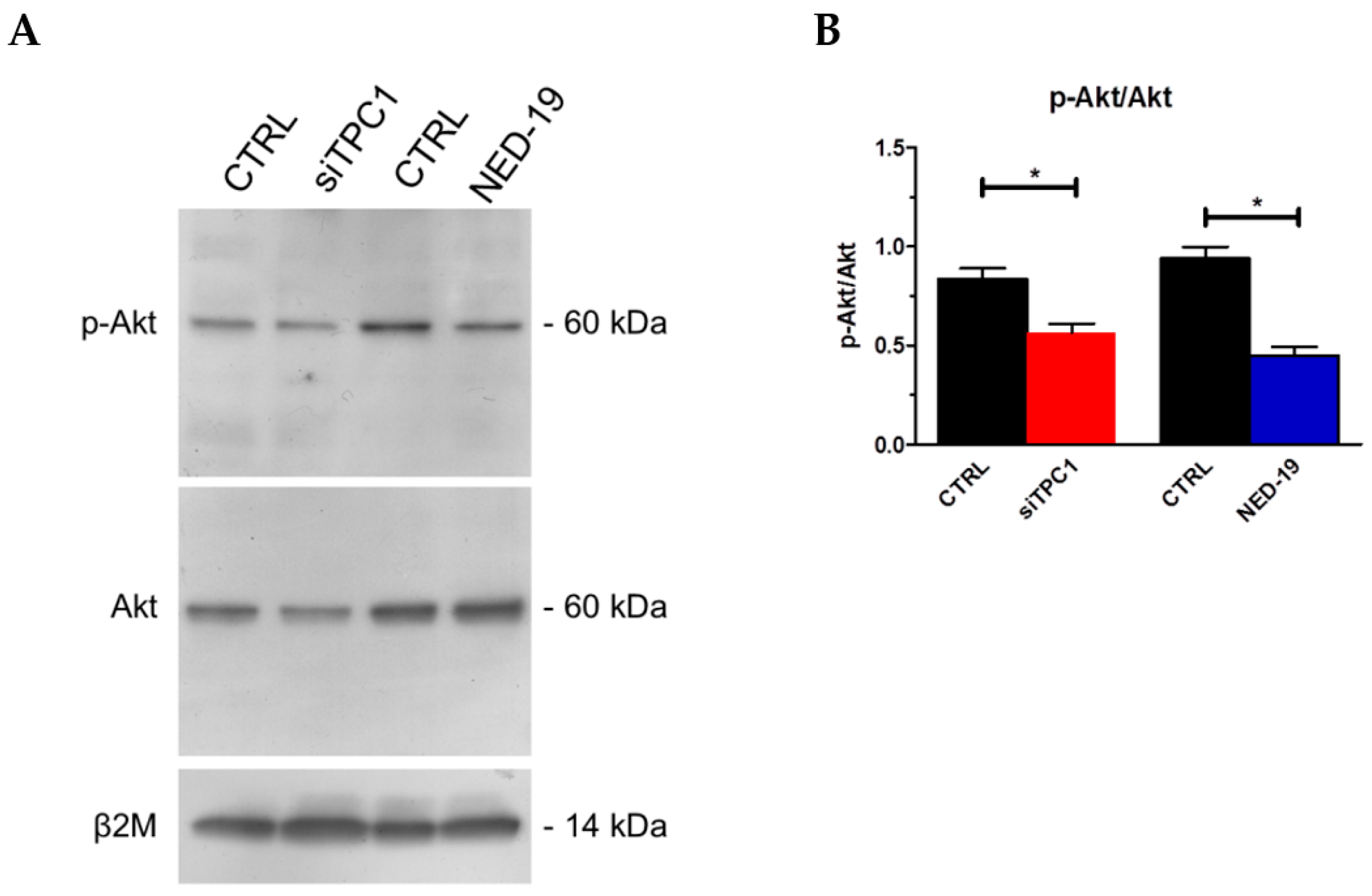
2.5. TPC1 Triggers FBS-Induced Intracellular Ca2+ Release and Stimulates Proliferation in Metastatic Colorectal Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Expansion of Tumor Cells
4.2. Solutions for Intracellular Ca2+ Recordings
4.3. Preparation of NAADP-Containing Liposomes
4.4. [Ca2+]i Measurements
4.5. RNA Isolation and Real Time RT-PCR (qRT-PCR)
4.6. Membrane Preparation and Immunoblotting
4.7. Protein Content
4.8. Gene Silencing
4.9. Lysotracker Red Staining
4.10. Statistics
4.11. Chemicals
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Samanta, K.; Parekh, A.B. Spatial Ca(2+) profiling: Decrypting the universal cytosolic Ca(2+) oscillation. J. Physiol. 2017, 595, 3053–3062. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal cell-derived factor-1alpha promotes endothelial colony-forming cell migration through the Ca(2+)-dependent activation of the extracellular signal-regulated kinase 1/2 and phosphoinositide 3-kinase/AKT pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef]
- Cardenas, C.; Muller, M.; McNeal, A.; Lovy, A.; Jana, F.; Bustos, G.; Urra, F.; Smith, N.; Molgo, J.; Diehl, J.A.; et al. Selective vulnerability of cancer cells by inhibition of Ca(2+) transfer from endoplasmic reticulum to mitochondria. Cell Rep. 2016, 15, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels in cancer: Are cancer hallmarks oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.K.; Galione, A. The endoplasmic reticulum and junctional membrane communication during calcium signaling. Biochim. Biophys. Acta 2013, 1833, 2542–2559. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef]
- Missiaen, L.; Dode, L.; Vanoevelen, J.; Raeymaekers, L.; Wuytack, F. Calcium in the Golgi apparatus. Cell Calcium 2007, 41, 405–416. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Galione, A. A primer of NAADP-mediated Ca(2+) signalling: From sea urchin eggs to mammalian cells. Cell Calcium 2015, 58, 27–47. [Google Scholar] [CrossRef]
- Morgan, A.J. Ca2+ dialogue between acidic vesicles and ER. Biochem. Soc. Trans. 2016, 44, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Melchionda, M.; Pittman, J.K.; Mayor, R.; Patel, S. Ca2+/H+ exchange by acidic organelles regulates cell migration in vivo. J. Cell Biol. 2016, 212, 803–813. [Google Scholar] [CrossRef]
- Ronco, V.; Potenza, D.M.; Denti, F.; Vullo, S.; Gagliano, G.; Tognolina, M.; Guerra, G.; Pinton, P.; Genazzani, A.A.; Mapelli, L.; et al. A novel Ca(2)(+)-mediated cross-talk between endoplasmic reticulum and acidic organelles: Implications for NAADP-dependent Ca(2)(+) signalling. Cell Calcium 2015, 57, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Z.; Gu, M.; Feng, X.; Xu, H. Release and uptake mechanisms of vesicular Ca(2+) stores. Protein Cell 2019, 10, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Schieder, M.; Cuny, H.; Fenske, S.; Gruner, C.; Rotzer, K.; Griesbeck, O.; Harz, H.; Biel, M.; Wahl-Schott, C. The two-pore channel TPCN2 mediates NAADP-dependent Ca(2+)-release from lysosomal stores. Pflug. Arch. 2009, 458, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Ruas, M.; Davis, L.C.; Chen, C.C.; Morgan, A.J.; Chuang, K.T.; Walseth, T.F.; Grimm, C.; Garnham, C.; Powell, T.; Platt, N.; et al. Expression of Ca(2)(+)-permeable two-pore channels rescues NAADP signalling in TPC-deficient cells. EMBO J. 2015, 34, 1743–1758. [Google Scholar] [CrossRef]
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H. Linking NAADP to ion channel activity: A unifying hypothesis. Sci. Signal. 2012, 5, pe18. [Google Scholar] [CrossRef]
- Wolf, I.M.; Diercks, B.P.; Gattkowski, E.; Czarniak, F.; Kempski, J.; Werner, R.; Schetelig, D.; Mittrucker, H.W.; Schumacher, V.; von Osten, M.; et al. Frontrunners of T cell activation: Initial, localized Ca2+ signals mediated by NAADP and the type 1 ryanodine receptor. Sci. Signal. 2015, 8, ra102. [Google Scholar] [CrossRef]
- Penny, C.J.; Kilpatrick, B.S.; Eden, E.R.; Patel, S. Coupling acidic organelles with the ER through Ca(2)(+) microdomains at membrane contact sites. Cell Calcium 2015, 58, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Ruas, M.; Rietdorf, K.; Arredouani, A.; Davis, L.C.; Lloyd-Evans, E.; Koegel, H.; Funnell, T.M.; Morgan, A.J.; Ward, J.A.; Watanabe, K.; et al. Purified TPC isoforms form NAADP receptors with distinct roles for Ca(2+) signaling and endolysosomal trafficking. Curr. Biol. 2010, 20, 703–709. [Google Scholar] [CrossRef]
- Moccia, F.; Nusco, G.A.; Lim, D.; Kyozuka, K.; Santella, L. NAADP and InsP3 play distinct roles at fertilization in starfish oocytes. Dev. Biol. 2006, 294, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vascul. Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca(2+) signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef]
- Brailoiu, E.; Hoard, J.L.; Filipeanu, C.M.; Brailoiu, G.C.; Dun, S.L.; Patel, S.; Dun, N.J. Nicotinic acid adenine dinucleotide phosphate potentiates neurite outgrowth. J. Biol. Chem. 2005, 280, 5646–5650. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.J.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Smaili, S.S. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef]
- Arredouani, A.; Ruas, M.; Collins, S.C.; Parkesh, R.; Clough, F.; Pillinger, T.; Coltart, G.; Rietdorf, K.; Royle, A.; Johnson, P.; et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) and endolysosomal two-pore channels modulate membrane excitability and stimulus-secretion coupling in mouse pancreatic beta cells. J. Biol. Chem. 2015, 290, 21376–21392. [Google Scholar] [CrossRef]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca(2+) oscillations and nitric oxide release by inducing NAADP-and InsP3-dependent Ca(2+) release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef]
- Patel, S.; Kilpatrick, B.S. Two-pore channels and disease. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1678–1686. [Google Scholar] [CrossRef]
- Sterea, A.M.; Almasi, S.; El Hiani, Y. The hidden potential of lysosomal ion channels: A new era of oncogenes. Cell Calcium 2018, 72, 91–103. [Google Scholar] [CrossRef]
- Grimm, C.; Bartel, K.; Vollmar, A.M.; Biel, M. Endolysosomal cation channels and cancer-a link with great potential. Pharmaceuticals (Basel) 2018, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Jahidin, A.H.; Stewart, T.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Differential effects of two-pore channel protein 1 and 2 silencing in MDA-MB-468 breast cancer cells. Biochem. Biophys. Res. Commun. 2016, 477, 731–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, O.N.; Grimm, C.; Schneider, L.S.; Chao, Y.K.; Atzberger, C.; Bartel, K.; Watermann, A.; Ulrich, M.; Mayr, D.; Wahl-Schott, C.; et al. Two-pore channel function is crucial for the migration of invasive cancer cells. Cancer Res. 2017, 77, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Pafumi, I.; Desideri, M.; Padula, F.; Montesano, C.; Passeri, D.; Nicoletti, C.; Orlandi, A.; Del Bufalo, D.; Sergi, M.; et al. NAADP-dependent Ca(2+) signaling controls melanoma progression, metastatic dissemination and neoangiogenesis. Sci. Rep. 2016, 6, 18925. [Google Scholar] [CrossRef]
- Turin, I.; Schiavo, R.; Maestri, M.; Luinetti, O.; Dal Bello, B.; Paulli, M.; Dionigi, P.; Roccio, M.; Spinillo, A.; Ferulli, F.; et al. In vitro efficient expansion of tumor cells deriving from different types of human tumor samples. Med. Sci. 2014, 2, 70–81. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai mediate constitutive Ca(2+) entry and control endoplasmic reticulum Ca(2+) refilling in primary cultures of colorectal carcinoma cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Davis, L.C.; Wagner, S.K.; Lewis, A.M.; Parrington, J.; Churchill, G.C.; Galione, A. Bidirectional Ca(2)(+) signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 2013, 200, 789–805. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Schapira, A.H.; Futter, C.E.; Patel, S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci. 2013, 126, 60–66. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, L.; Bardo, S.J.; Emptage, N.J. The lysosome or lysosome-related organelle may serve as a Ca2+ store in the boutons of hippocampal pyramidal cells. Neuropharmacology 2007, 52, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Chuang, C.C.; Lewis, A.M.; Aley, P.K.; Brailoiu, E.; Dun, N.J.; Churchill, G.C.; Patel, S. Recruitment of NAADP-sensitive acidic Ca2+ stores by glutamate. Biochem. J. 2009, 422, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Cancela, J.M.; Van Coppenolle, F.; Galione, A.; Tepikin, A.V.; Petersen, O.H. Transformation of local Ca2+ spikes to global Ca2+ transients: The combinatorial roles of multiple Ca2+ releasing messengers. EMBO J. 2002, 21, 909–919. [Google Scholar] [CrossRef]
- Penny, C.J.; Kilpatrick, B.S.; Han, J.M.; Sneyd, J.; Patel, S. A computational model of lysosome-ER Ca2+ microdomains. J. Cell Sci. 2014, 127, 2934–2943. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting Stim and Orai proteins as an alternative approach in anticancer therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Di Nezza, F.; Zuccolo, E.; Poletto, V.; Rosti, V.; De Luca, A.; Moccia, F.; Guerra, G.; Ambrosone, L. Liposomes as a putative tool to investigate NAADP signaling in vasculogenesis. J. Cell. Biochem. 2017, 118, 3722–3729. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Bong, A.H.; Poo, G.X.H.; Armitage, K.; Lok, D.; Roberts-Thomson, S.J.; Monteith, G.R. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal A breast cancer cells: Consequences on calcium signalling, cell migration and proliferation. Cell. Mol. Life Sci. 2018, 75, 4525–4537. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [PubMed]
- Kinnear, N.P.; Wyatt, C.N.; Clark, J.H.; Calcraft, P.J.; Fleischer, S.; Jeyakumar, L.H.; Nixon, G.F.; Evans, A.M. Lysosomes co-localize with ryanodine receptor subtype 3 to form a trigger zone for calcium signalling by NAADP in rat pulmonary arterial smooth muscle. Cell Calcium 2008, 44, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelu, J.J.; Chan, H.L.; Webb, S.E.; Cheng, A.H.; Ruas, M.; Parrington, J.; Galione, A.; Miller, A.L. Two-pore channel 2 activity is required for slow muscle cell-generated Ca(2+) signaling during myogenesis in intact zebrafish. Int. J. Dev. Biol. 2015, 59, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Pitt, S.J.; Reilly-O’Donnell, B.; Sitsapesan, R. Exploring the biophysical evidence that mammalian two-pore channels are NAADP-activated calcium-permeable channels. J. Physiol. 2016, 594, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.C.; Morgan, A.J.; Chen, J.L.; Snead, C.M.; Bloor-Young, D.; Shenderov, E.; Stanton-Humphreys, M.N.; Conway, S.J.; Churchill, G.C.; Parrington, J.; et al. NAADP activates two-pore channels on T cell cytolytic granules to stimulate exocytosis and killing. Curr. Biol. 2012, 22, 2331–2337. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.L.; Lin, A.H.; Xia, Y.; Lee, S.; Paudel, O.; Sun, H.; Yang, X.R.; Ran, P.; Sham, J.S. Nicotinic acid adenine dinucleotide phosphate (NAADP) activates global and heterogeneous local Ca2+ signals from NAADP-and ryanodine receptor-gated Ca2+ stores in pulmonary arterial myocytes. J. Biol. Chem. 2013, 288, 10381–10394. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef]
- Moccia, F. Endothelial Ca(2+) Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef]
- Moccia, F.; Poletto, V. May the remodeling of the Ca(2)(+) toolkit in endothelial progenitor cells derived from cancer patients suggest alternative targets for anti-angiogenic treatment? Biochim. Biophys. Acta 2015, 1853, 1958–1973. [Google Scholar] [CrossRef]
- Pinto, M.C.; Kihara, A.H.; Goulart, V.A.; Tonelli, F.M.; Gomes, K.N.; Ulrich, H.; Resende, R.R. Calcium signaling and cell proliferation. Cell Signal. 2015, 27, 2139–2149. [Google Scholar] [CrossRef]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Carrato, A. Adjuvant treatment of colorectal cancer. Gastrointest. Cancer Res. 2008, 2 (4 Suppl.), S42–S46. [Google Scholar] [PubMed]
- Turin, I.; Delfanti, S.; Ferulli, F.; Brugnatelli, S.; Tanzi, M.; Maestri, M.; Cobianchi, L.; Lisini, D.; Luinetti, O.; Paulli, M.; et al. In vitro killing of colorectal carcinoma cells by autologous activated NK cells is boosted by anti-epidermal growth factor receptor-induced ADCC regardless of RAS mutation status. J. Immunother. 2018, 41, 190–200. [Google Scholar] [CrossRef]
- Bufalo, G.; Di Nezza, F.; Cimino, L.; Cuomo, F.; Ambrosone, L. Physicochemical investigation of ultrasound effects on some steps of mink fur processing. A suggestion for improving the worker health and reducing the environmental impact. J. Clean. Prod. 2017, 143, 10–16. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Spano, A.; Barni, S.; Bertone, V.; Sciola, L. Changes on lysosomal compartment during PMA-induced differentiation of THP-1 monocytic cells: Influence of type I and type IV collagens. Adv. Biosci. Biotechnol. 2013, 4, 8–18. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faris, P.; Pellavio, G.; Ferulli, F.; Di Nezza, F.; Shekha, M.; Lim, D.; Maestri, M.; Guerra, G.; Ambrosone, L.; Pedrazzoli, P.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca2+ Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells. Cancers 2019, 11, 542. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11040542
Faris P, Pellavio G, Ferulli F, Di Nezza F, Shekha M, Lim D, Maestri M, Guerra G, Ambrosone L, Pedrazzoli P, et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca2+ Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells. Cancers. 2019; 11(4):542. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11040542
Chicago/Turabian StyleFaris, Pawan, Giorgia Pellavio, Federica Ferulli, Francesca Di Nezza, Mudhir Shekha, Dmitry Lim, Marcello Maestri, Germano Guerra, Luigi Ambrosone, Paolo Pedrazzoli, and et al. 2019. "Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca2+ Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells" Cancers 11, no. 4: 542. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11040542