The Actin Binding Protein Plastin-3 Is Involved in the Pathogenesis of Acute Myeloid Leukemia

Abstract
:1. Introduction
2. Results
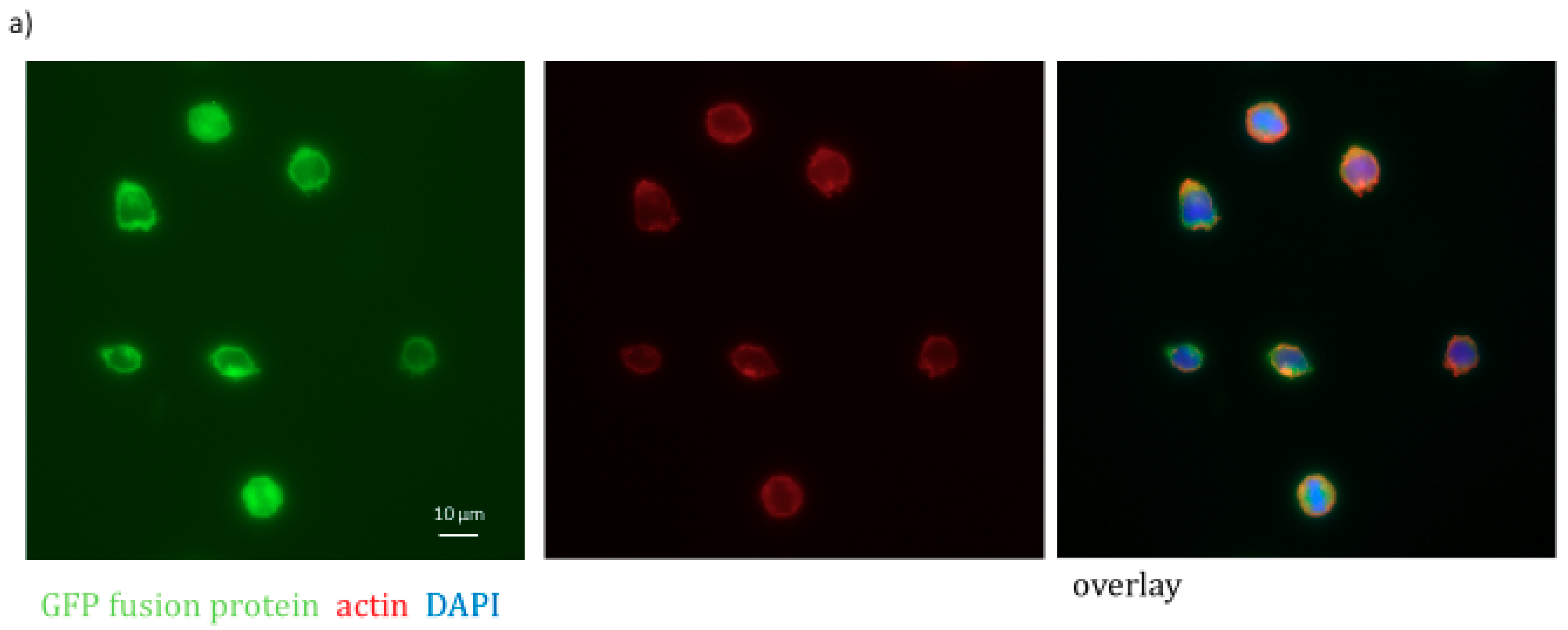
2.1. The Actin Binding Protein PLS3 Is Expressed in Primary AML Cells
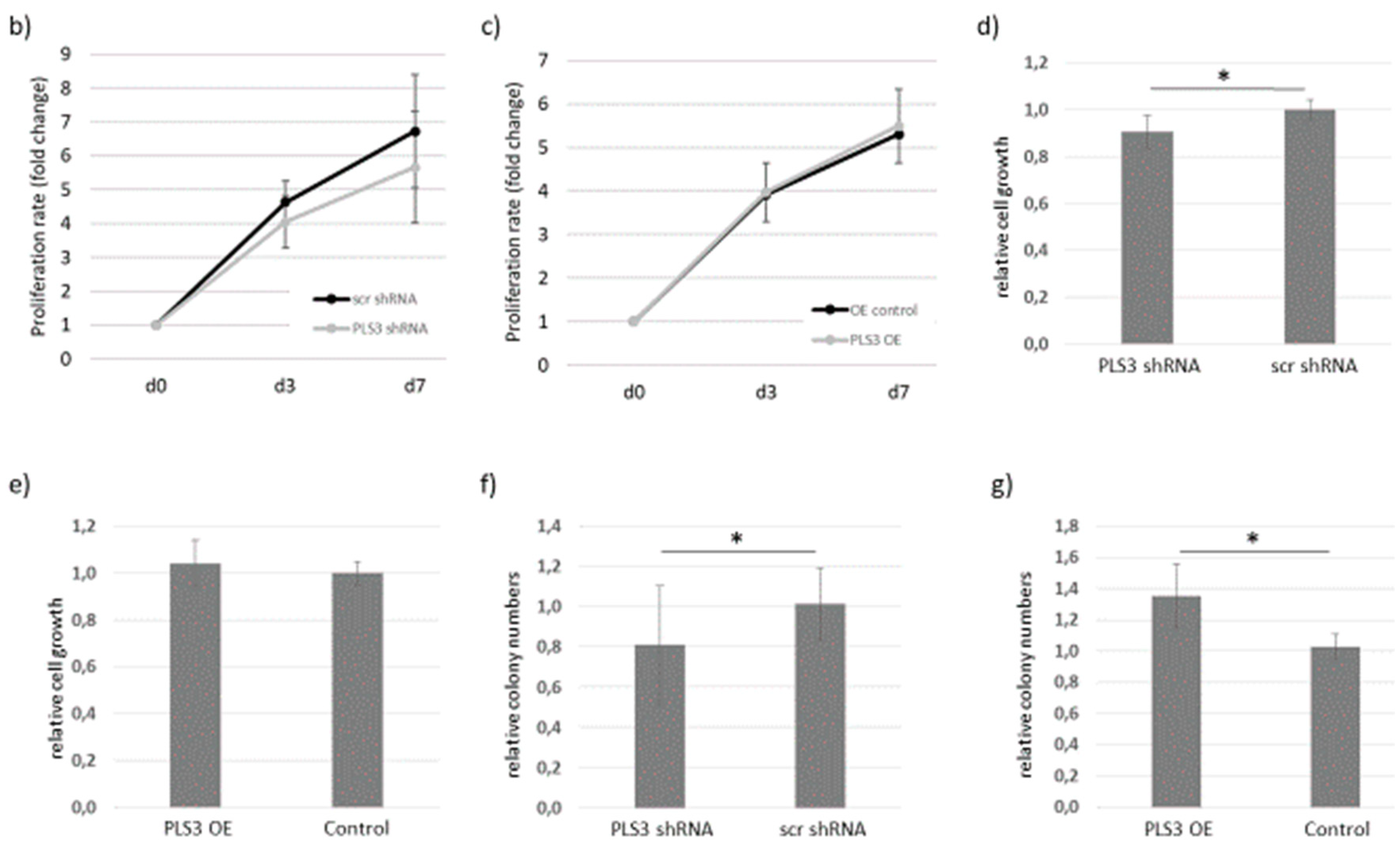
2.2. Functional Analysis of PSL3 by Knockdown or Overexpression in Kasumi-1 Cells
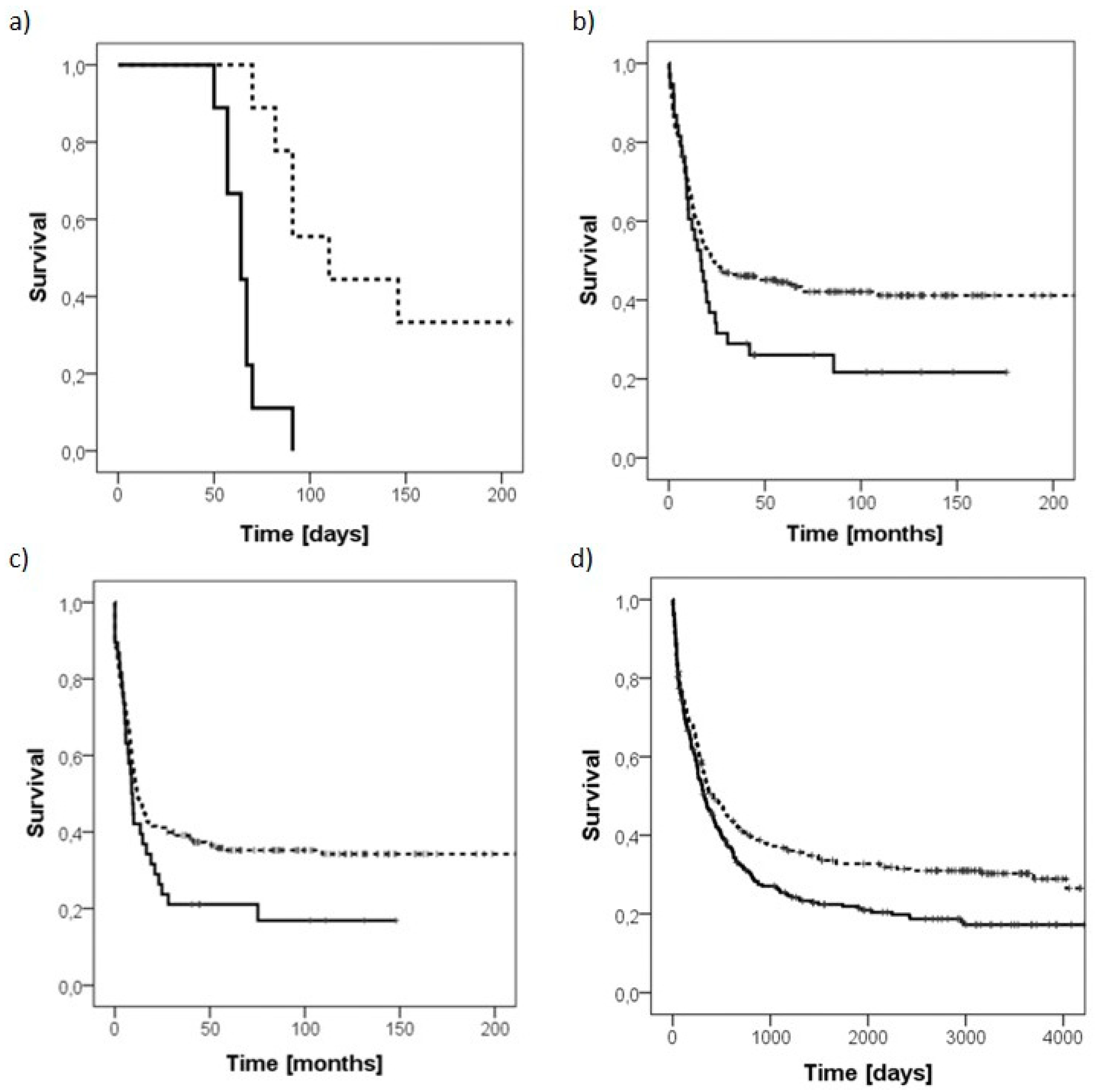
2.3. Knockdown of PLS3 Significantly Improves Survival In Vivo
2.4. PLS3 Represents a Negative Prognostic Marker in AML
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Cell Culture
4.3. Reverse Transcription Quantitative PCR Analysis
4.4. Immunofluorescence Staining
4.5. Lentiviral Transduction of Kasumi-1 Cells with PLS3-Specific shRNA or a PLS3 Overexpression Construct
4.6. Staining of F-Actin
4.7. Proliferation Assays
4.8. Colony Formation Assay
4.9. Xenograft Model
4.10. RNA Sequencing
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Cogle, C.R.; Bosse, R.C.; Brewer, T.; Migdady, Y.; Shirzad, R.; Kampen, K.R.; Saki, N. Acute myeloid leukemia in the vascular niche. Cancer Lett. 2016, 380, 552–560. [Google Scholar] [CrossRef]
- Delanote, V.; Vandekerckhove, J.; Gettemans, J. Plastins: Versatile modulators of actin organization in (patho)physiological cellular processes. Acta Pharmacol. Sin. 2005, 26, 769–779. [Google Scholar] [CrossRef]
- Lin, C.S.; Aebersold, R.H.; Kent, S.B.; Varma, M.; Leavitt, J. Molecular cloning and characterization of plastin, a human leukocyte protein expressed in transformed human fibroblasts. Mol. Cell. Biol. 1988, 8, 4659–4668. [Google Scholar] [CrossRef]
- Giganti, A.; Plastino, J.; Janji, B.; Van Troys, M.; Lentz, D.; Ampe, C.; Sykes, C.; Friederich, E. Actin-filament cross-linking protein T-plastin increases Arp2/3-mediated actin-based movement. J. Cell Sci. 2005, 118, 1255–1265. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Wouters, B.J.; Erpelinck, C.A.J.; Abbas, S.; Beverloo, H.B.; Lugthart, S.; Lowenberg, B.; Delwel, R.; Valk, P.J.M. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica 2009, 94, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Herold, T.; He, C.; Valk, P.J.M.; Chen, P.; Jurinovic, V.; Mansmann, U.; Radmacher, M.D.; Maharry, K.S.; Sun, M.; et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: An international collaborative study. J. Clin. Oncol. 2013, 31, 1172–1181. [Google Scholar] [CrossRef]
- Jones, C.L.; Ferreira, S.; McKenzie, R.C.T.; Tosi, I.; Caesar, J.A.; Bagot, M.; Whittaker, S.J.; Mitchell, T.J. Regulation of T-plastin expression by promoter hypomethylation in primary cutaneous T-cell lymphoma. J. Investig. Dermatol. 2012, 132, 2042–2049. [Google Scholar] [CrossRef]
- Henn, A.; Michel, L.; Fite, C.; Deschamps, L.; Ortonne, N.; Ingen-Housz-Oro, S.; Marinho, E.; Beylot-Barry, M.; Bagot, M.; Laroche, L.; et al. Sézary syndrome without erythroderma. J. Am. Acad. Dermatol. 2015, 72, 1003–1009.e1. [Google Scholar] [CrossRef] [PubMed]
- Kurashige, J.; Yokobori, T.; Mima, K.; Sawada, G.; Takahashi, Y.; Ueo, H.; Takano, Y.; Matsumura, T.; Uchi, R.; Eguchi, H.; et al. Plastin3 is associated with epithelial-mesenchymal transition and poor prognosis in gastric cancer. Oncol. Lett. 2019, 17, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Szkandera, J.; Winder, T.; Stotz, M.; Weissmueller, M.; Langsenlehner, T.; Pichler, M.; Samonigg, H.; Renner, W.; Gerger, A.; Absenger, G. A common gene variant in PLS3 predicts colon cancer recurrence in women. Tumour Biol. 2013, 34, 2183–2188. [Google Scholar] [CrossRef]
- Yokobori, T.; Iinuma, H.; Shimamura, T.; Imoto, S.; Sugimachi, K.; Ishii, H.; Iwatsuki, M.; Ota, D.; Ohkuma, M.; Iwaya, T.; et al. Plastin3 is a novel marker for circulating tumor cells undergoing the epithelial-mesenchymal transition and is associated with colorectal cancer prognosis. Cancer Res. 2013, 73, 2059–2069. [Google Scholar] [CrossRef] [PubMed]
- Kujawski, R.; Przybyłowska-Sygut, K.; Mik, M.; Lewandowski, M.; Trzciński, R.; Berut, M.; Dziki, Ł.; Majsterek, I.; Dziki, A. Expression of the PLS3 Gene in Circulating Cells in Patients with Colorectal Cancer. Pol. Przegl. Chir. 2015, 87, 59–64. [Google Scholar] [CrossRef]
- Ueo, H.; Sugimachi, K.; Gorges, T.M.; Bartkowiak, K.; Yokobori, T.; Müller, V.; Shinden, Y.; Ueda, M.; Ueo, H.; Mori, M.; et al. Circulating tumour cell-derived plastin3 is a novel marker for predicting long-term prognosis in patients with breast cancer. Br. J. Cancer 2015, 112, 1519–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Lai, W.; Zhao, M.; Yue, C.; Shi, F.; Li, R.; Hu, Z. Plastin 3 down-regulation augments the sensitivity of MDA-MB-231 cells to paclitaxel via the p38 MAPK signalling pathway. Artif. Cells Nanomed. Biotechnol. 2019, 47, 685–695. [Google Scholar] [CrossRef]
- Hisano, T.; Ono, M.; Nakayama, M.; Naito, S.; Kuwano, M.; Wada, M. Increased expression of T-plastin gene in cisplatin-resistant human cancer cells: Identification by mRNA differential display. FEBS Lett. 1996, 397, 101–107. [Google Scholar] [CrossRef]
- Higuchi, Y.; Kita, K.; Nakanishi, H.; Wang, X.L.; Sugaya, S.; Tanzawa, H.; Yamamori, H.; Sugita, K.; Yamaura, A.; Suzuki, N. Search for genes involved in UV-resistance in human cells by mRNA differential display: Increased transcriptional expression of nucleophosmin and T-plastin genes in association with the resistance. Biochem. Biophys. Res. Commun. 1998, 248, 597–602. [Google Scholar] [CrossRef]
- Sasaki, Y.; Itoh, F.; Kobayashi, T.; Kikuchi, T.; Suzuki, H.; Toyota, M.; Imai, K. Increased expression of T-fimbrin gene after DNA damage in CHO cells and inactivation of T-fimbrin by CpG methylation in human colorectal cancer cells. Int. J. Cancer 2002, 97, 211–216. [Google Scholar] [CrossRef]
- Neugebauer, J.; Heilig, J.; Hosseinibarkooie, S.; Ross, B.C.; Mendoza-Ferreira, N.; Nolte, F.; Peters, M.; Hölker, I.; Hupperich, K.; Tschanz, T.; et al. Plastin 3 influences bone homeostasis through regulation of osteoclast activity. Hum. Mol. Genet. 2018, 27, 4249–4262. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, F.A.; Pierce, J.W.; Sonenshein, G.E. Differential regulation of the c-myc oncogene promoter by the NF-kappa B rel family of transcription factors. Mol. Cell. Biol. 1994, 14, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ma, L.; Li, J.; Yu, Y.; Zhang, D.; Wei, J.; Jin, H.; Xu, D.; Gao, J.; Huang, C. NF-κB1 inhibits c-Myc protein degradation through suppression of FBW7 expression. Oncotarget 2014, 5, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Dang, C. V MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Ohanian, M.; Rozovski, U.; Kanagal-Shamanna, R.; Abruzzo, L.V.; Loghavi, S.; Kadia, T.; Futreal, A.; Bhalla, K.; Zuo, Z.; Huh, Y.O.; et al. MYC protein expression is an important prognostic factor in acute myeloid leukemia. Leuk. Lymphoma 2019, 60, 37–48. [Google Scholar] [CrossRef]
- Vecchione, A.; Ishii, H.; Baldassarre, G.; Bassi, P.; Trapasso, F.; Alder, H.; Pagano, F.; Gomella, L.G.; Croce, C.M.; Baffa, R. FEZ1/LZTS1 is down-regulated in high-grade bladder cancer, and its restoration suppresses tumorigenicity in transitional cell carcinoma cells. Am. J. Pathol. 2002, 160, 1345–1352. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Z.; Sun, X.; Dong, X.-Y.; Wei, J.; Gu, F.; Sun, Y.-L.; Zhou, J.; Dong, J.-T.; Fu, L. Down-regulation of tumor suppressor gene FEZ1/LZTS1 in breast carcinoma involves promoter methylation and associates with metastasis. Breast Cancer Res. Treat. 2009, 116, 471–478. [Google Scholar] [CrossRef]
- Toyooka, S.; Fukuyama, Y.; Wistuba, I.I.; Tockman, M.S.; Minna, J.D.; Gazdar, A.F. Differential expression of FEZ1/LZTS1 gene in lung cancers and their cell cultures. Clin. Cancer Res. 2002, 8, 2292–2297. [Google Scholar]
- Lanza, D.C.F.; Trindade, D.M.; Assmann, E.M.; Kobarg, J. Over-expression of GFP-FEZ1 causes generation of multi-lobulated nuclei mediated by microtubules in HEK293 cells. Exp. Cell Res. 2008, 314, 2028–2039. [Google Scholar] [CrossRef]
- Bertini Teixeira, M.; Figueira, A.C.M.; Furlan, A.S.; Aquino, B.; Alborghetti, M.R.; Paes Leme, A.F.; Wei, L.-N.; Kobarg, J. Fasciculation and elongation zeta-1 protein (FEZ1) interacts with the retinoic acid receptor and participates in transcriptional regulation of the Hoxb4 gene. FEBS Open Bio 2018, 8, 4–14. [Google Scholar] [CrossRef]
- Zhang, X.-B.; Beard, B.C.; Trobridge, G.D.; Wood, B.L.; Sale, G.E.; Sud, R.; Humphries, R.K.; Kiem, H.-P. High incidence of leukemia in large animals after stem cell gene therapy with a HOXB4-expressing retroviral vector. J. Clin. Investig. 2008, 118, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Bodey, B.; Bodey, B.; Siegel, S.E.; Kaiser, H.E. Immunocytochemical detection of the homeobox B3, B4, and C6 gene products in breast carcinomas. Anticancer Res. 2000, 20, 3281–3286. [Google Scholar] [PubMed]
- Wang, H.; Jia, X.-H.; Chen, J.-R.; Yi, Y.-J.; Wang, J.-Y.; Li, Y.-J.; Xie, S.-Y. HOXB4 knockdown reverses multidrug resistance of human myelogenous leukemia K562/ADM cells by downregulating P-gp, MRP1 and BCRP expression via PI3K/Akt signaling pathway. Int. J. Oncol. 2016, 49, 2529–2537. [Google Scholar] [CrossRef] [PubMed]
- Jeimy, S.B.; Tasneem, S.; Cramer, E.M.; Hayward, C.P.M. Multimerin 1. Platelets 2008, 19, 83–95. [Google Scholar] [CrossRef]
- Laszlo, G.S.; Alonzo, T.A.; Gudgeon, C.J.; Harrington, K.H.; Gerbing, R.B.; Wang, Y.-C.; Ries, R.E.; Raimondi, S.C.; Hirsch, B.A.; Gamis, A.S.; et al. Multimerin-1 (MMRN1) as Novel Adverse Marker in Pediatric Acute Myeloid Leukemia: A Report from the Children’s Oncology Group. Clin. Cancer Res. 2015, 21, 3187–3195. [Google Scholar] [CrossRef]
- Xu, Z.; Xi, K. LncRNA RGMB-AS1 promotes laryngeal squamous cell carcinoma cells progression via sponging miR-22/NLRP3 axis. Biomed. Pharmacother. 2019, 118, 109222. [Google Scholar] [CrossRef]
- Li, P.; Zhang, G.; Li, J.; Yang, R.; Chen, S.; Wu, S.; Zhang, F.; Bai, Y.; Zhao, H.; Wang, Y.; et al. Long Noncoding RNA RGMB-AS1 Indicates a Poor Prognosis and Modulates Cell Proliferation, Migration and Invasion in Lung Adenocarcinoma. PLoS ONE 2016, 11, e0150790. [Google Scholar] [CrossRef]
- Sheng, N.; Li, Y.; Qian, R.; Li, Y. The clinical significance and biological function of lncRNA RGMB-AS1 in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 98, 577–584. [Google Scholar] [CrossRef]
- Löwenberg, B.; Boogaerts, M.A.; Daenen, S.M.; Verhoef, G.E.; Hagenbeek, A.; Vellenga, E.; Ossenkoppele, G.J.; Huijgens, P.C.; Verdonck, L.F.; van der Lelie, J.; et al. Value of different modalities of granulocyte-macrophage colony-stimulating factor applied during or after induction therapy of acute myeloid leukemia. J. Clin. Oncol. 1997, 15, 3496–3506. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FAB Subtype | PLS3 (%) | ||
|---|---|---|---|
| Low | High | Fisher’s Exact Test | |
| M1 (n = 67) | 81 | 19 | p = 0.077 |
| M2 (n = 64) | 94 | 6 | |
| M4 (n = 59) | 93 | 7 | |
| M5 (n = 62) | 97 | 13 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velthaus, A.; Cornils, K.; Hennigs, J.K.; Grüb, S.; Stamm, H.; Wicklein, D.; Bokemeyer, C.; Heuser, M.; Windhorst, S.; Fiedler, W.; et al. The Actin Binding Protein Plastin-3 Is Involved in the Pathogenesis of Acute Myeloid Leukemia. Cancers 2019, 11, 1663. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111663
Velthaus A, Cornils K, Hennigs JK, Grüb S, Stamm H, Wicklein D, Bokemeyer C, Heuser M, Windhorst S, Fiedler W, et al. The Actin Binding Protein Plastin-3 Is Involved in the Pathogenesis of Acute Myeloid Leukemia. Cancers. 2019; 11(11):1663. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111663
Chicago/Turabian StyleVelthaus, Arne, Kerstin Cornils, Jan K. Hennigs, Saskia Grüb, Hauke Stamm, Daniel Wicklein, Carsten Bokemeyer, Michael Heuser, Sabine Windhorst, Walter Fiedler, and et al. 2019. "The Actin Binding Protein Plastin-3 Is Involved in the Pathogenesis of Acute Myeloid Leukemia" Cancers 11, no. 11: 1663. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111663