WRN-Mutated Colorectal Cancer Is Characterized by a Distinct Genetic Phenotype


 , , ,
, , ,
Abstract
:1. Introduction
2. Results
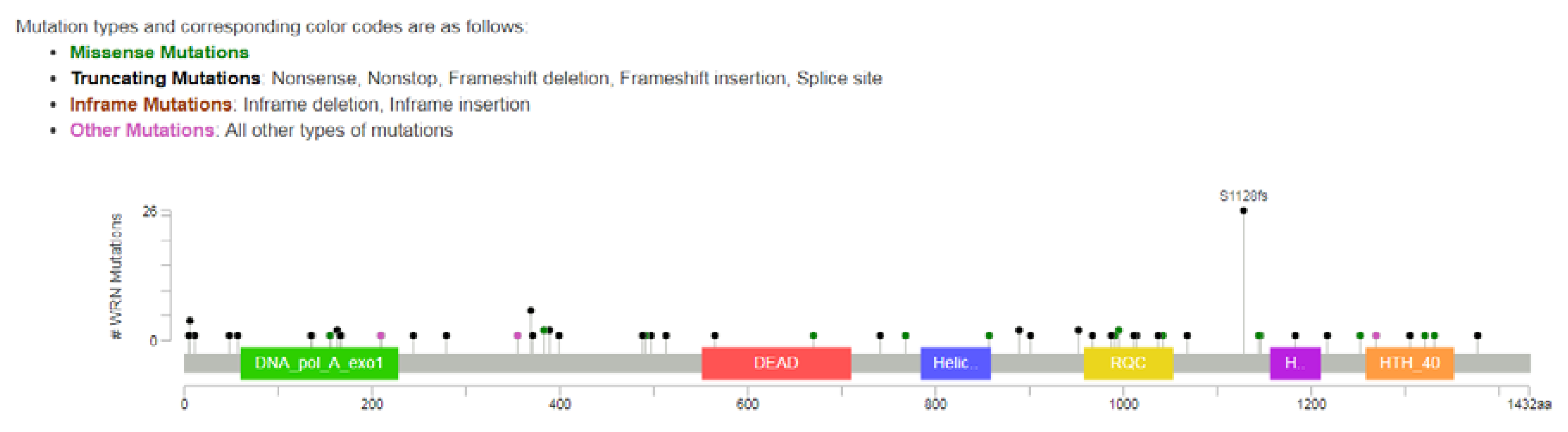
2.1. Incidence of WRN Mutations in Colorectal Cancer
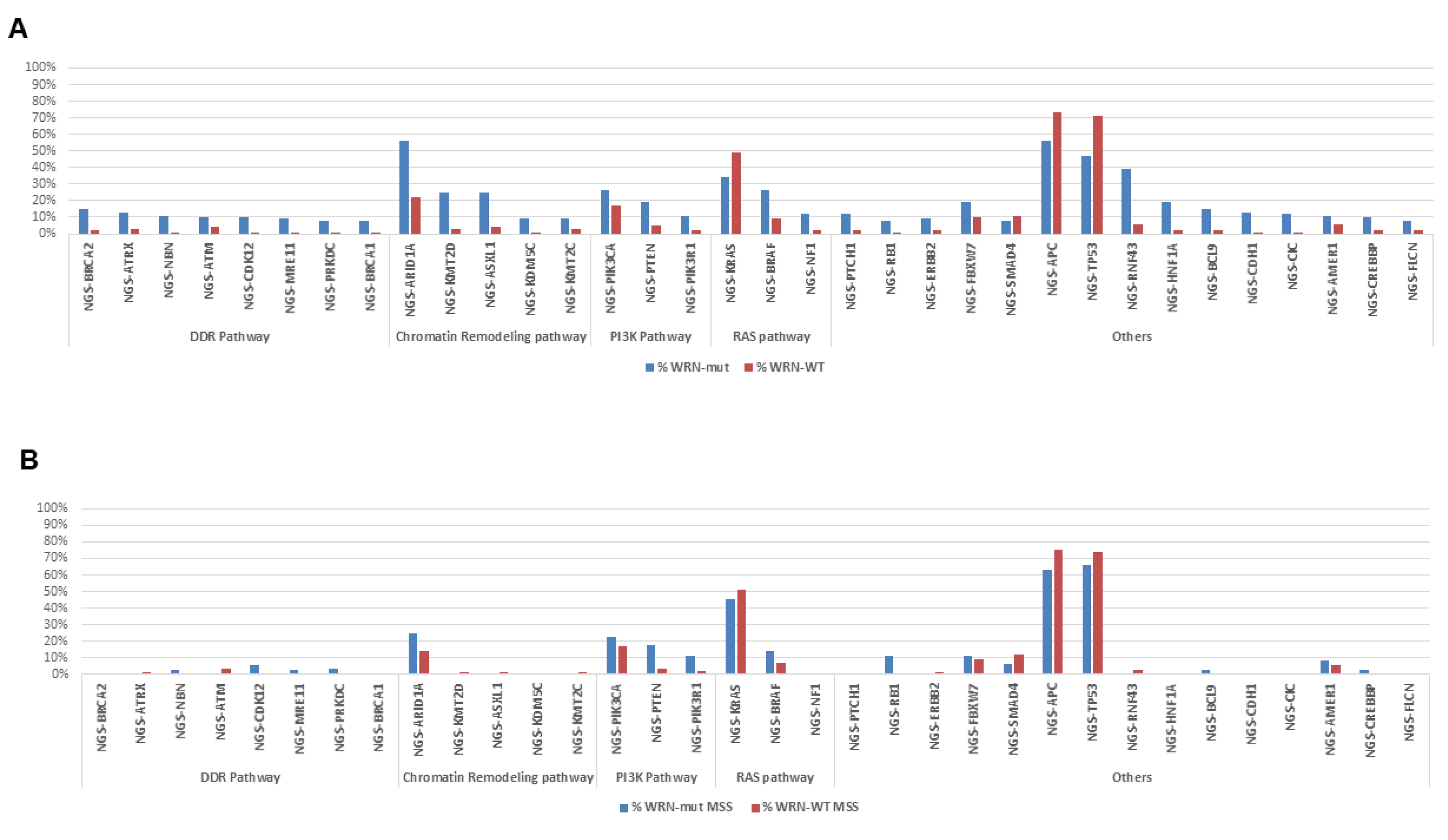
2.2. Molecular Portrait of WRN-Mutated and Wild-Type Colorectal Cancer
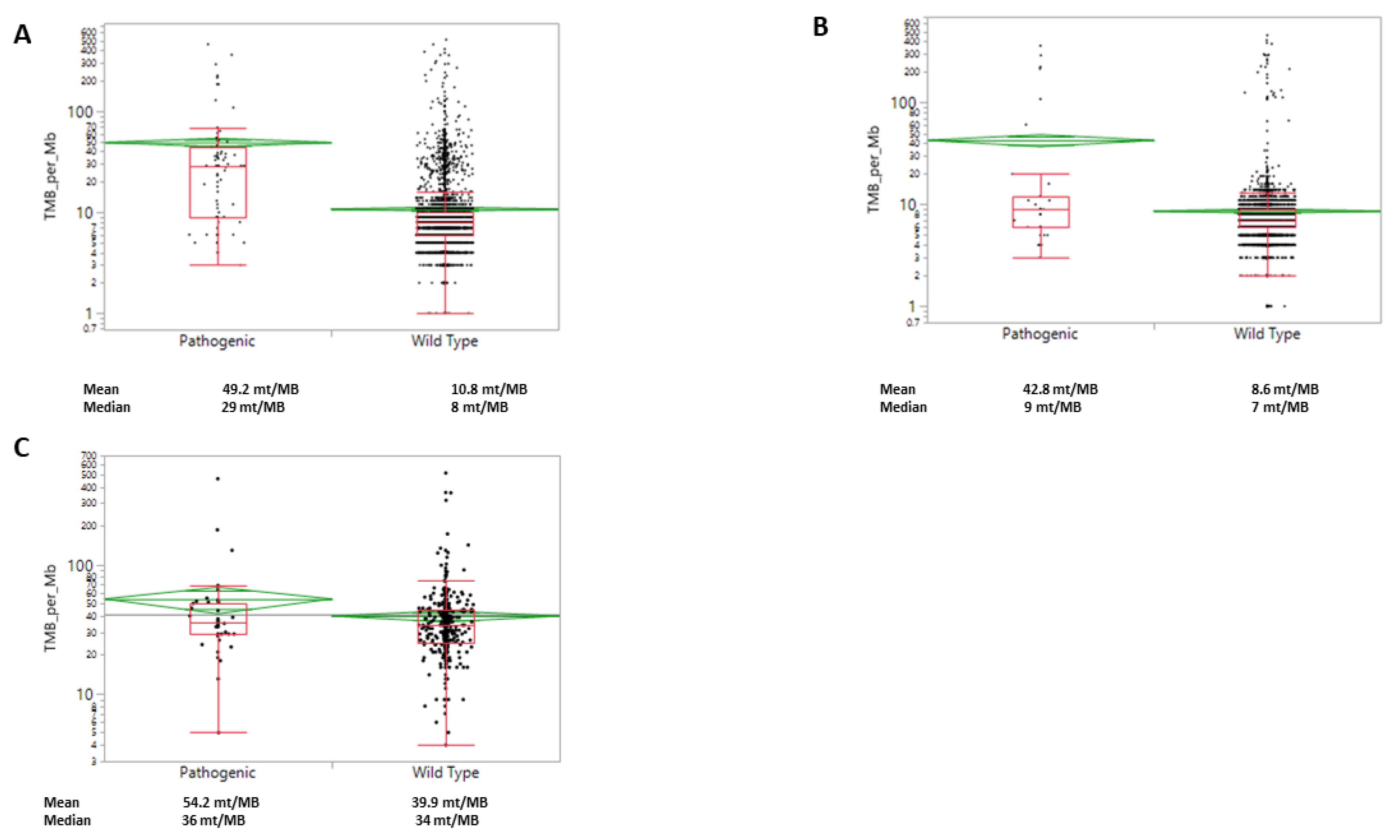
2.3. WRN Mutations Correlate with PDL1, TMB and MSI-H/dMMR
3. Discussion
4. Material and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fitzmaurice, C. Global Burden of Disease Cancer Collaboration Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 2006 to 2016: A systematic analysis for the Global Burden of Disease study. J. Clin. Oncol. 2018, 36, 1568. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; Sileni, V.C.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.-J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients With Advanced Non‒Small-Cell Lung Cancer Treated With Pembrolizumab: Results From the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Venderbosch, S.; Nagtegaal, I.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.D.; et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN and FOCUS studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- Koopman, M.; Kortman, G.A.M.; Mekenkamp, L.; Ligtenberg, M.J.L.; Hoogerbrugge, N.; Antonini, N.F.; A Punt, C.J.; Van Krieken, J.H.J.M. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Dudley, J.C.; Lin, M.-T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Chan, E.; Shibue, T.; McFarland, J.M.; Gaeta, B.; Ghandi, M.; Dumont, N.; Gonzalez, A.; McPartlan, J.S.; Li, T.; Zhang, Y.; et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 2019, 568, 551–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Lauper, J.M.; Krause, A.; Vaughan, T.L.; Monnat, R.J. Spectrum and Risk of Neoplasia in Werner Syndrome: A Systematic Review. PLoS ONE 2013, 8, e59709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, J.; Sidorova, J.M.; Monnat, R.J. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 2016, 33, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lao, V.; Carter, K.; Dzieciatkowski, S.; Welcsh, P.; Rabinovich, P.; Sarvetnick, N.; Grady, W. Altered RecQ Helicase Expression in Sporadic Primary Colorectal Cancers. J. Surg. Res. 2013, 179, 340. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, M.; Morikawa, T.; Kuchiba, A.; Imamura, Y.; Qian, Z.R.; Nishihara, R.; Liao, X.; Waldron, L.; Hoshida, Y.; Huttenhower, C.; et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 2012, 61, 847–854. [Google Scholar] [CrossRef]
- Baran, B.; Ozupek, N.M.; Tetik, N.Y.; Acar, E.; Bekcioglu, O.; Baskin, Y. Difference Between Left-Sided and Right-Sided Colorectal Cancer: A Focused Review of Literature. Gastroenterol. Res. 2018, 11, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterol. 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.M.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Leichman, L.; Groshen, S.; O’Neil, B.H.; Messersmith, W.; Berlin, J.; Chan, E.; Leichman, C.G.; Cohen, S.J.; Cohen, D.; Lenz, H.-J.; et al. Phase II Study of Olaparib (AZD-2281) After Standard Systemic Therapies for Disseminated Colorectal Cancer. Oncologist 2016, 21, 172–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Jonker, D.J.; Siu, L.L.; McKeever, K.; Keller, D.; Wells, J.; Hagerman, L.; Seymour, L. A Phase I study of olaparib and irinotecan in patients with colorectal cancer: Canadian Cancer Trials Group IND 187. Investig. New Drugs 2016, 34, 450–457. [Google Scholar] [CrossRef]
- Kawasaki, T.; Ohnishi, M.; Suemoto, Y.; Kirkner, G.J.; Liu, Z.; Yamamoto, H.; Loda, M.; Fuchs, C.S.; Ogino, S. WRN promoter methylation possibly connects mucinous differentiation, microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod. Pathol. 2007, 21, 150–158. [Google Scholar] [CrossRef]
- Prelaj, A.; Tay, R.; Ferrara, R.; Chaput, N.; Besse, B.; Califano, R. Predictive biomarkers of response for immune checkpoint inhibitors in non-small-cell lung cancer. Eur. J. Cancer 2018, 106, 144–159. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Schrock, A.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.; Miller, V.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [Green Version]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Giannakis, M.; Hodis, E.; Mu, X.J.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Seeber, A.; Kocher, F.; Xiu, J.; Spizzo, G.; Puccini, A.; Swensen, J.; Ellis, M.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; et al. Molecular landscape of colorectal cancers harboring R-spondin fusions. J. Clin. Oncol. 2019, 37, 3588. [Google Scholar] [CrossRef]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Banerjee, T.; A Sommers, J.; Brosh, J.R.M. Targeting an Achilles’ heel of cancer with a WRN helicase inhibitor. Function of a membrane-embedded domain evolutionarily multiplied in the GPI lipid anchor pathway proteins PIG-B, PIG-M, PIG-U, PIG-W, PIG-V, and PIG-Z. Cell Cycle 2013, 12, 3329–3335. [Google Scholar] [CrossRef]
- Nguyen, G.H.; Dexheimer, T.S.; Rosenthal, A.S.; Chu, W.K.; Singh, D.K.; Mosedale, G.; Bachrati, C.Z.; Schultz, L.; Sakurai, M.; Savitsky, P.; et al. A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem. Boil. 2013, 20, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Sommers, J.A.; Kulikowicz, T.; Croteau, D.L.; Dexheimer, T.; Dorjsuren, D.; Jadhav, A.; Maloney, D.J.; Simeonov, A.; Bohr, V.A.; Brosh, R.M. A high-throughput screen to identify novel small molecule inhibitors of the Werner Syndrome Helicase-Nuclease (WRN). PLoS ONE 2019, 14, e0210525. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Brosh, R.M. New Insights Into DNA Helicases as Druggable Targets for Cancer Therapy. Front. Mol. Biosci. 2018, 5, 59. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WRN—No Mutation | WRN—Mutation | Mutation % | p Value | ||
|---|---|---|---|---|---|
| Specimen Type | Metastasis | 2921 | 28 | 0.9% | 0.0034 |
| Primary/local | 3853 | 52 | 1.3% | ||
| Total | 6774 | 80 | 1.2% | – | |
| Age | Median age | 60.3 | 62.5 | – | NS (not significant) |
| Gender | Female | 3062 | 38 | 1.2% | NS |
| Male | 3712 | 42 | 1.1% | ||
| Sidedness | Left | 3371 | 24 | 0.7% | p < 0.0001 |
| Right | 1743 | 44 | 2.5% | ||
| NOS (not otherwise specified) | 1660 | 12 | 0.7% | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmer, K.; Puccini, A.; Xiu, J.; Baca, Y.; Spizzo, G.; Lenz, H.-J.; Battaglin, F.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; et al. WRN-Mutated Colorectal Cancer Is Characterized by a Distinct Genetic Phenotype. Cancers 2020, 12, 1319. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051319
Zimmer K, Puccini A, Xiu J, Baca Y, Spizzo G, Lenz H-J, Battaglin F, Goldberg RM, Grothey A, Shields AF, et al. WRN-Mutated Colorectal Cancer Is Characterized by a Distinct Genetic Phenotype. Cancers. 2020; 12(5):1319. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051319
Chicago/Turabian StyleZimmer, Kai, Alberto Puccini, Joanne Xiu, Yasmine Baca, Gilbert Spizzo, Heinz-Josef Lenz, Francesca Battaglin, Richard M. Goldberg, Axel Grothey, Anthony F. Shields, and et al. 2020. "WRN-Mutated Colorectal Cancer Is Characterized by a Distinct Genetic Phenotype" Cancers 12, no. 5: 1319. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051319