Aqueous Dehydration, Hydrogenation, and Hydrodeoxygenation Reactions of Bio-Based Mucic Acid over Ni, NiMo, Pt, Rh, and Ru on Neutral or Acidic Catalyst Supports


Abstract
:
1. Introduction
2. Results and Discussion
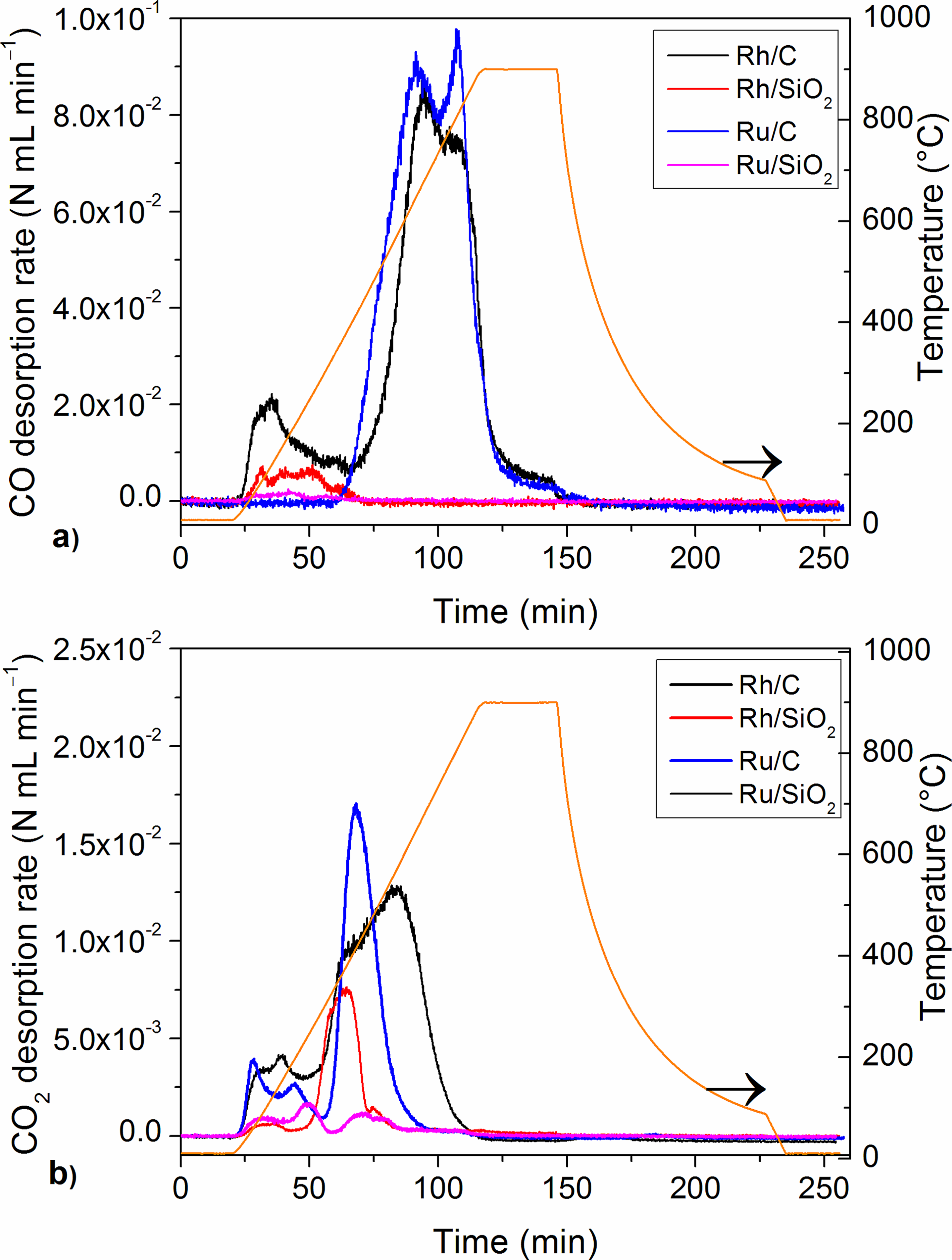
2.1. Catalyst Characterization
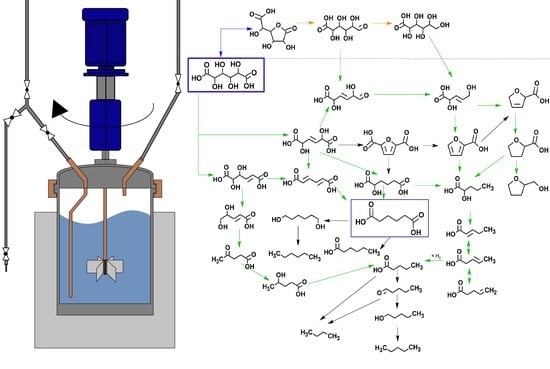
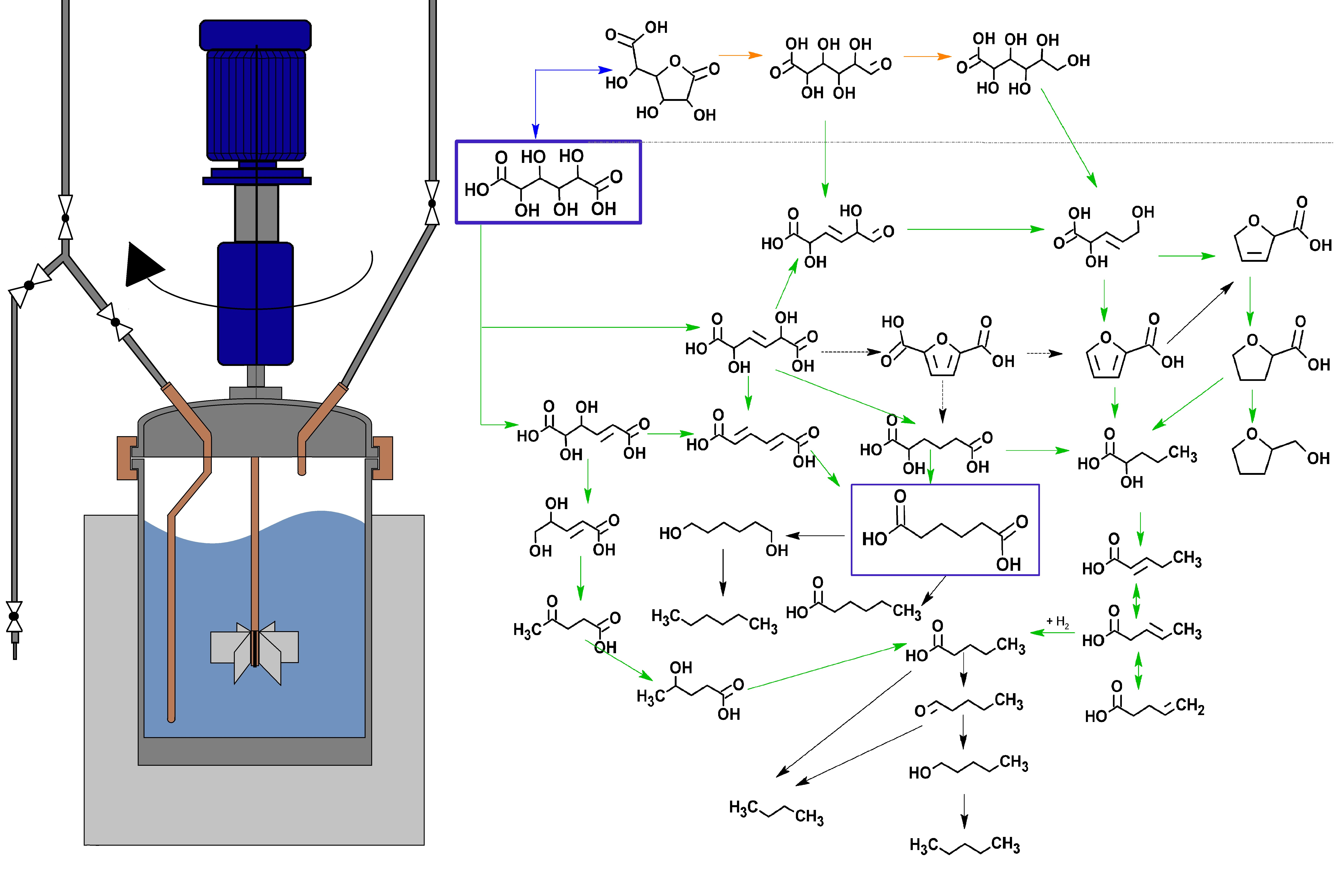
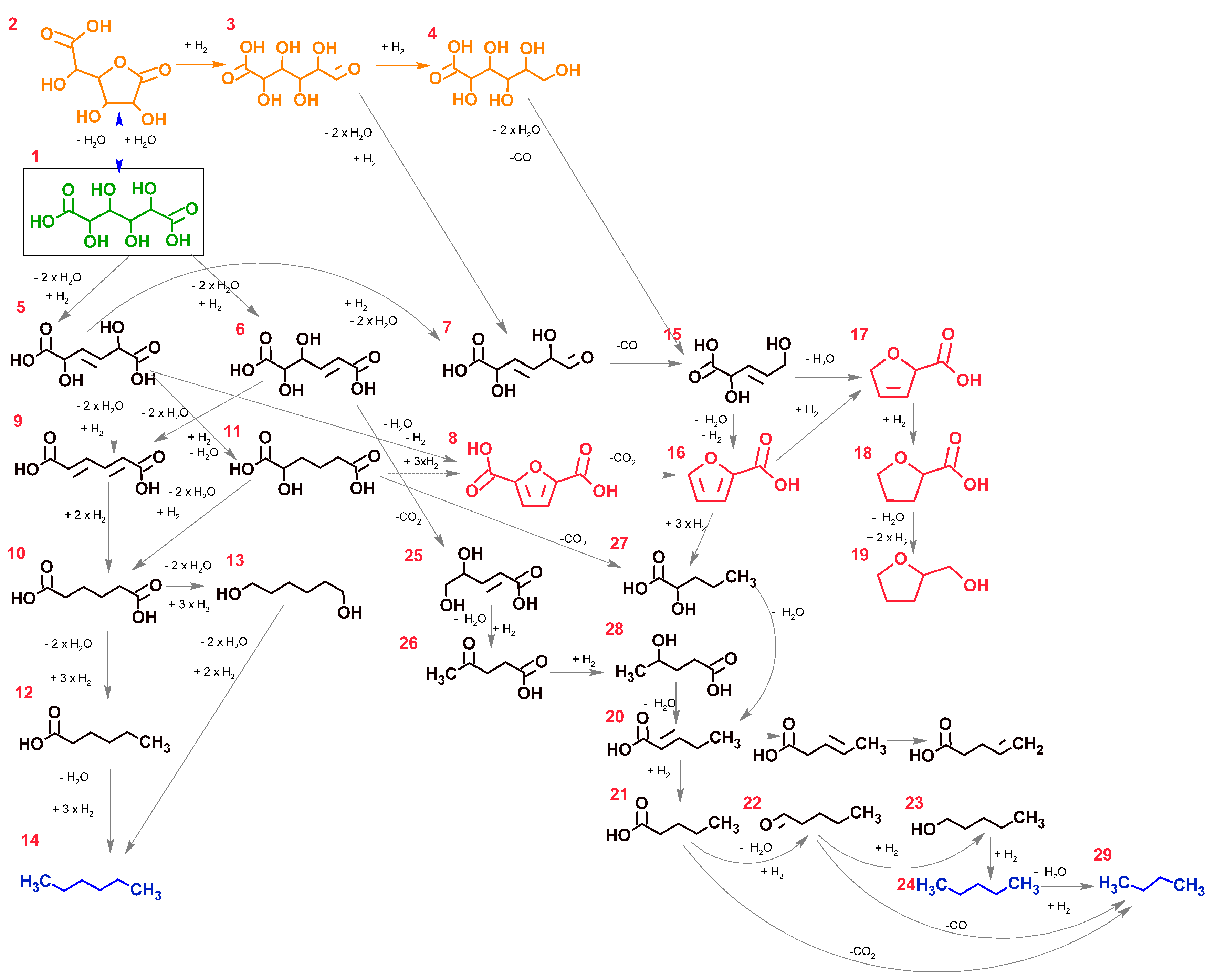
2.2. Reaction Pathway Development: Bulk Phase Reactions
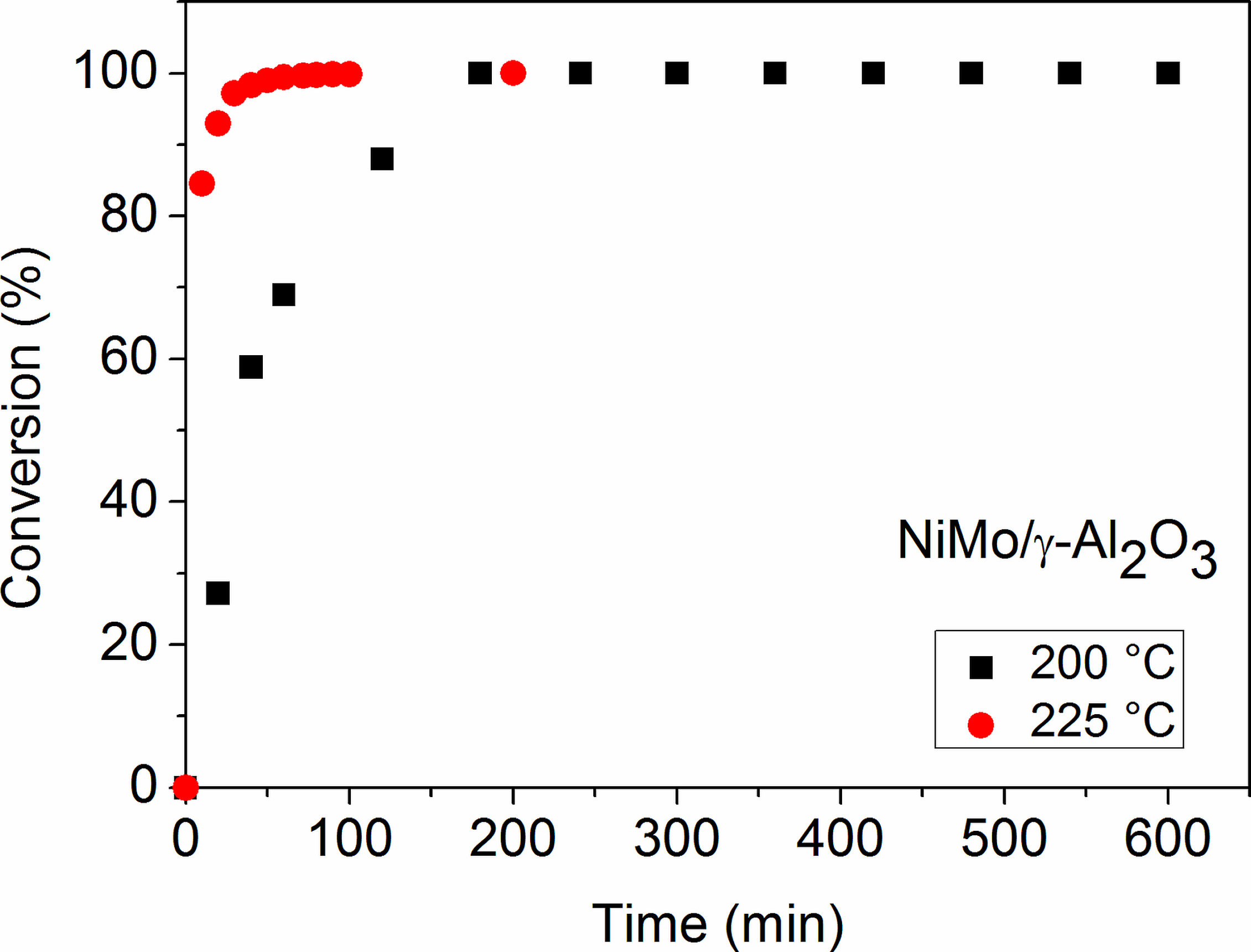
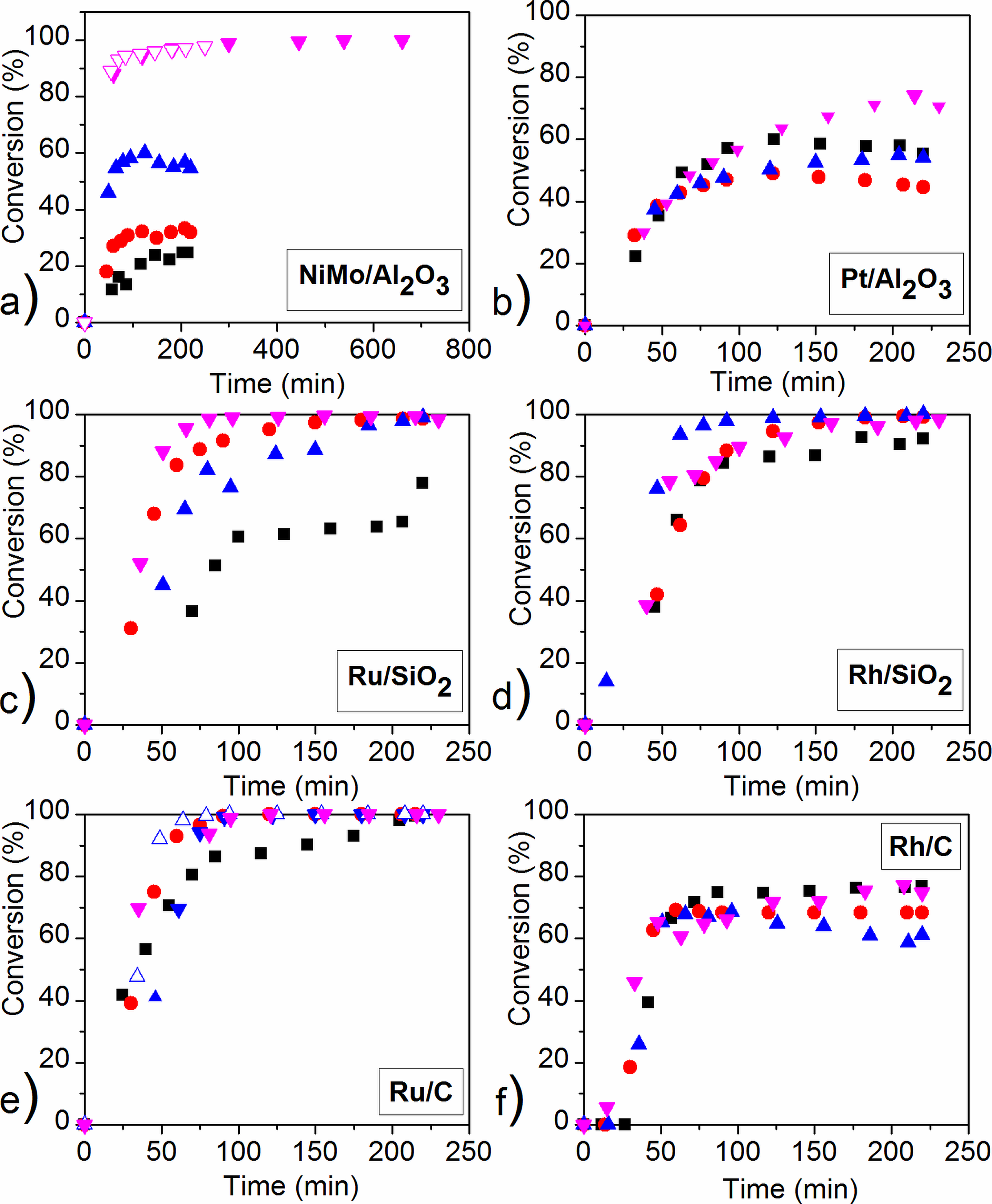
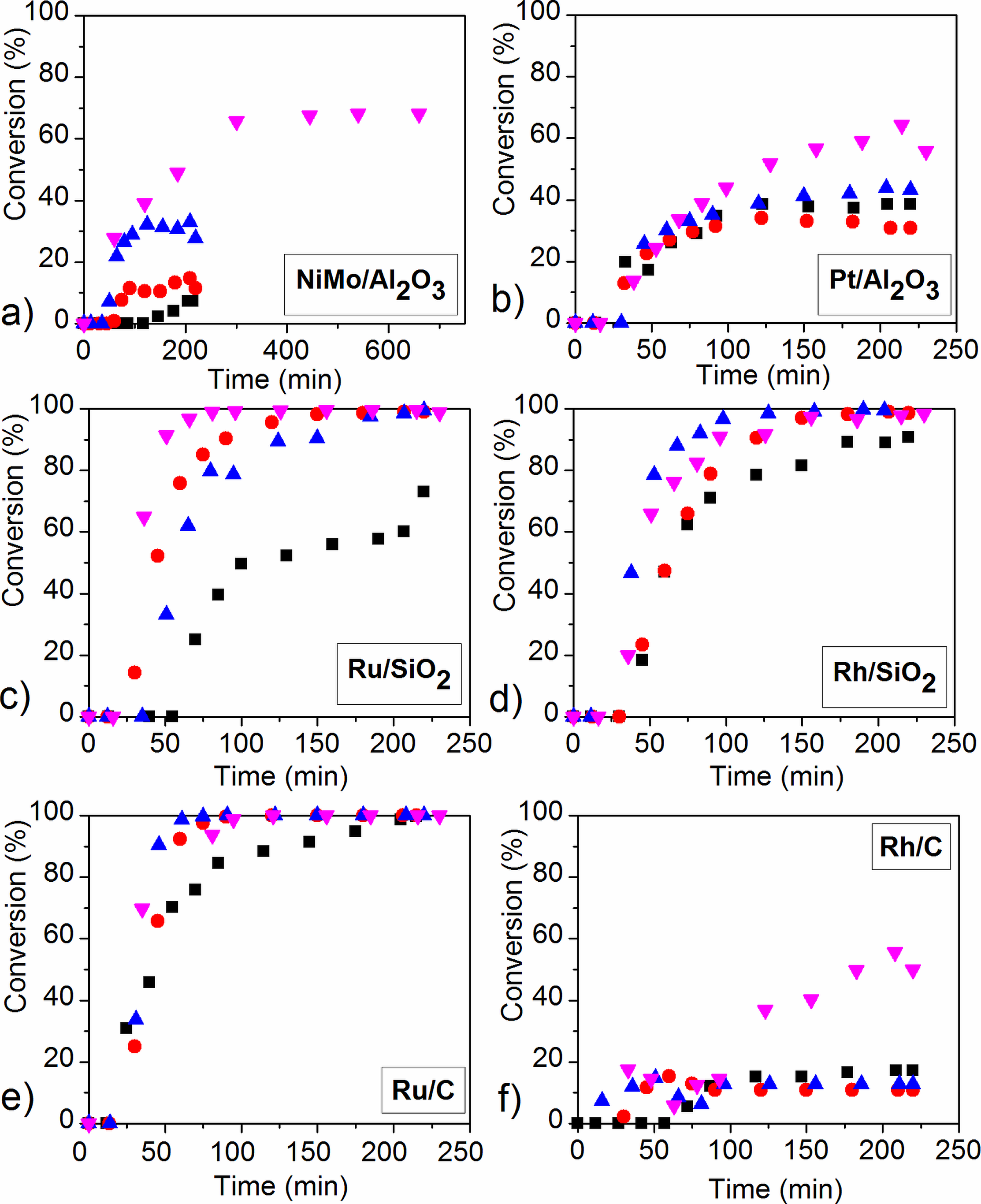
2.3. Reaction Pathway Development: Catalytic Reactions
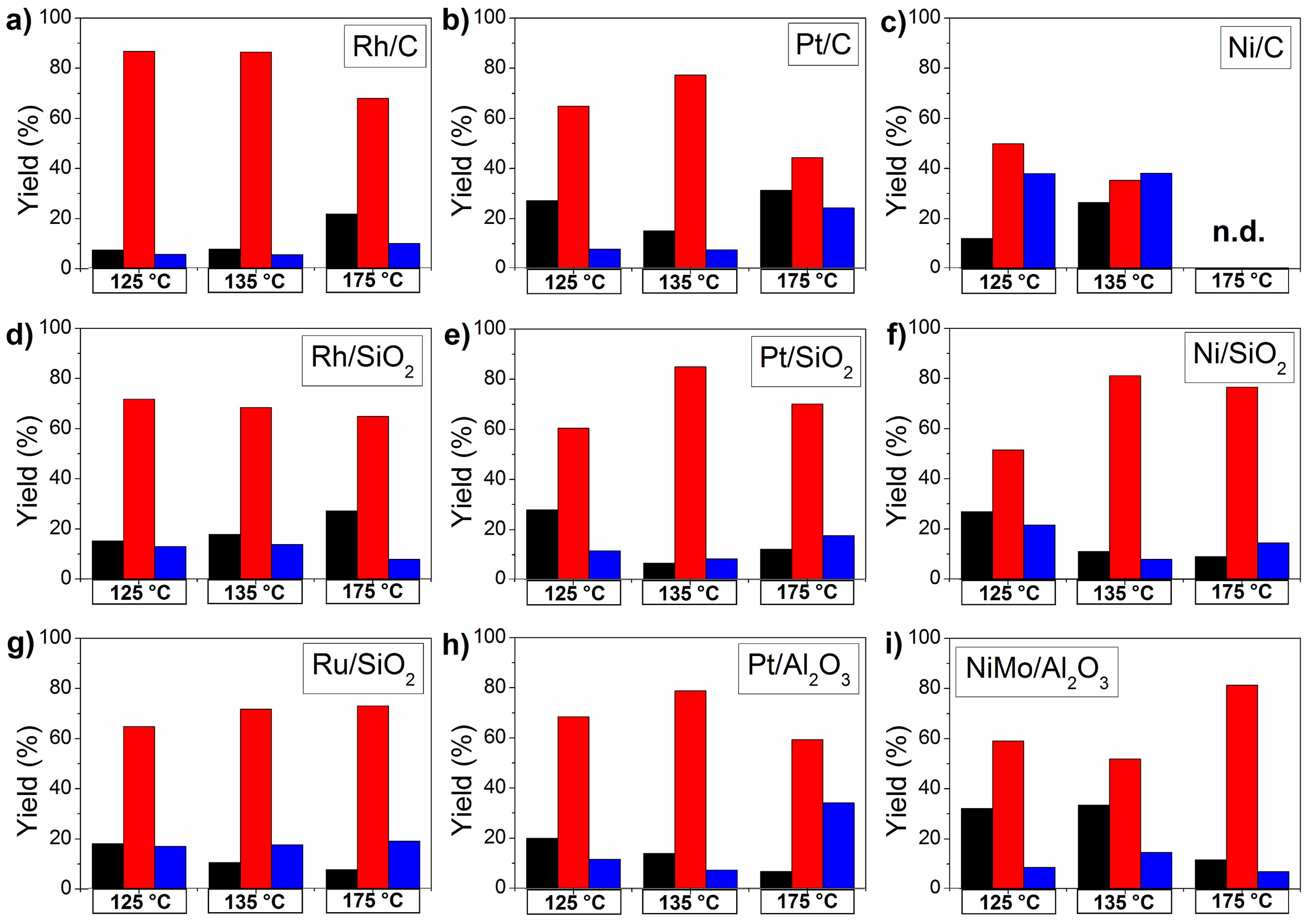
2.4. The Influence of Catalyst Type and Reaction Conditions on the HDO Selectivity of Mucic Acid
3. Materials and Methods
3.1. Catalyst Characterization
3.2. Hydrotreatment Experiments
3.3. Analytic Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavani, F. Chemicals and Fuels from Bio-based Building Blocks; Wiley-VCH Verlag GmbH & Company KGaA: Weinheim, Germany, 2016. [Google Scholar]
- Philp, J.C.; Ritchie, R.J.; Allan, J.E.M. Biobased chemicals: the convergence of green chemistry with industrial biotechnology. Trends Biotechnol. 2013, 31, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Werpy, T.; Petersen, G.; Aden, A.; Bozell, J.; Holladay, J.; White, J.; Manheim, A.; Eliot, D.; Lasure, L.; Jones, S. Top Value Added Chemicals from Biomass. Volume 1-Results of Screening for Potential Candidates from Sugars and Synthesis Gas; Department of Energy: Washington, DC, USA, 2004.
- Van Haveren, J.; Scott, E.L.; Sanders, J. Bulk chemicals from biomass. Biofuels Bioprod. Biorefining 2008, 2, 41–57. [Google Scholar] [CrossRef]
- Lederkremer, R.; Marino, C. Acids and Other Products of Oxidation of Sugars. Adv. Carbohydr. Chem. Biochem. 2004, 58, 199–306. [Google Scholar]
- Boussie, T.R.; Dias, E.L.; Fresco, Z.M.; Murphy, V.J.; Shoemaker, J.; Archer, R.; Jiang, H. Production of Adipic Acid and Derivatives from Carbohydrate-Containing Materials. U.S. Patent 8,501,989, 6 August 2010. [Google Scholar]
- Asikainen, M.; Thomas, D.; Harlin, A. Method for producing muconic acids and furans from aldaric acids. U.S. Patent 15/317,983, 18 May 2017. [Google Scholar]
- Yoshinao, N.; Sibao, L.; Masazumi, T.; Keiichi, T. Catalytic Total Hydrodeoxygenation of Biomass-Derived Polyfunctionalized Substrates to Alkanes. ChemSusChem 2015, 8, 1114–1132. [Google Scholar]
- Agblevor, F.A.; Jahromi, H. Aqueous-Phase Synthesis of Hydrocarbons from Furfural Reactions with Low-Molecular-Weight Biomass Oxygenates. Energy Fuels 2018, 32, 8552–8562. [Google Scholar] [CrossRef]
- Jahromi, H.; Agblevor, F.A. Hydrodeoxygenation of Aqueous-Phase Catalytic Pyrolysis Oil to Liquid Hydrocarbons Using Multifunctional Nickel Catalyst. Ind. Eng. Chem. Res. 2018, 57, 13257–13268. [Google Scholar] [CrossRef]
- Liang, C.; Donald, E. Kiely D-Glucaric Acid Esters/Lactones Used in Condensation Polymerization to Produce Hydroxylated Nylons—A Qualitative Equilibrium Study in Acidic and Basic Alcohol Solutions. J. Carbohydr. Chem. 1994, 13, 585. [Google Scholar]
- Si, Z.; Zhang, X.; Wang, C.; Ma, L.; Dong, R. An Overview on Catalytic Hydrodeoxygenation of Pyrolysis Oil and Its Model Compounds. Catalysts 2017, 7, 169. [Google Scholar] [CrossRef]
- Han, X.; Guo, Y.; Liu, X.; Xia, Q.; Wang, Y. Catalytic conversion of lignocellulosic biomass into hydrocarbons: a mini review. Catal. Today 2018. [Google Scholar] [CrossRef]
- Van Looij, F.; van der Laan, P.; Stork, W.H.J.; DiCamillo, D.J.; Swain, J. Key parameters in deep hydrodesulfurization of diesel fuel. Appl. Catal. A Gen. 1998, 170, 1–12. [Google Scholar] [CrossRef]
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Şenol, O.İ.; Ryymin, E.-M.; Viljava, T.-R.; Krause, A.O.I. Effect of hydrogen sulphide on the hydrodeoxygenation of aromatic and aliphatic oxygenates on sulphided catalysts. J. Mol. Catal. A Chem. 2007, 277, 107–112. [Google Scholar] [CrossRef]
- Boussie, T.R.; Diamond, D.M.; Dias, E.; Murphy, V. Synthesis of Adipic Acid Starting from Renewable Raw Materials. In Chemicals and Fuels from Bio-Based Building Blocks; Wiley-Blackwell: Hoboken, NJ, USA, 2016; pp. 151–172. ISBN 9783527698202. [Google Scholar]
- Yang, M.; Somorjai, G.A. Adsorption and Reactions of C6 Hydrocarbons at High Pressures on Pt(111) Single-Crystal Surfaces Studied by Sum Frequency Generation Vibrational Spectroscopy: Mechanisms of Isomerization and Dehydrocyclization of n-Hexane. J. Am. Chem. Soc. 2004, 126, 7698–7708. [Google Scholar] [CrossRef] [PubMed]
- Hočevar, B.; Grilc, M.; Huš, M.; Likozar, B. Mechanism, ab initio calculations and microkinetics of hydrogenation, hydrodeoxygenation, double bond migration and cis–trans isomerisation during hydrotreatment of {C6} secondary alcohol species and ketones. Appl. Catal. B Environ. 2017, 218, 147–162. [Google Scholar] [CrossRef]
- Hočevar, B.; Grilc, M.; Huš, M.; Likozar, B. Mechanism, ab initio calculations and microkinetics of straight-chain alcohol, ether, ester, aldehyde and carboxylic acid hydrodeoxygenation. Chem. Eng. J. 2019, 359, 1339–1351. [Google Scholar] [CrossRef]
- Grilc, M.; Likozar, B. Levulinic acid hydrodeoxygenation, decarboxylation and oligmerization over NiMo/Al2O3 catalyst to bio-based value-added chemicals: Modelling of mass transfer, thermodynamics and micro-kinetics. Chem. Eng. J. 2017, 330, 383–397. [Google Scholar] [CrossRef]
- Lefèvre, G.; Duc, M.; Lepeut, P.; Caplain, R.; Fédoroff, M. Hydration of γ-Alumina in Water and Its Effects on Surface Reactivity. Langmuir 2002, 18, 7530–7537. [Google Scholar] [CrossRef]
- Petek, U.; Ruiz-Zepeda, F.; Bele, M.; Gaberšček, M. Nanoparticles and Single Atoms in Commercial Carbon-Supported Platinum-Group Metal Catalysts. Catalysts 2019, 9, 134. [Google Scholar] [CrossRef]
- Tan, M.; Yang, G.; Wang, T.; Vitidsant, T.; Li, J.; Wei, Q.; Ai, P.; Wu, M.; Zheng, J.; Tsubaki, N. Active and regioselective rhodium catalyst supported on reduced graphene oxide for 1-hexene hydroformylation. Catal. Sci. Technol. 2016, 6, 1162–1172. [Google Scholar] [CrossRef]
- Linke, R.; Curulla, D.; Hopstaken, M.J.P.; Niemantsverdriet, J.W. CO/Rh(111): Vibrational frequency shifts and lateral interactions in adsorbate layers. J. Chem. Phys. 2001, 115, 8209–8216. [Google Scholar] [CrossRef]
- De Mongeot, F.B.; Scherer, M.; Gleich, B.; Kopatzki, E.; Behm, R.J. CO adsorption and oxidation on bimetallic Pt/Ru(0001) surfaces—A combined STM and TPD/TPR study. Surf. Sci. 1998, 411, 249–262. [Google Scholar] [CrossRef]
- LaRue, J.L.; Katayama, T.; Lindenberg, A.; Fisher, A.S.; Öström, H.; Nilsson, A.; Ogasawara, H. THz-Pulse-Induced Selective Catalytic CO Oxidation on Ru. Phys. Rev. Lett. 2015, 115, 36103. [Google Scholar] [CrossRef] [PubMed]
- Grenoble, D.C.; Estadt, M.M.; Ollis, D.F. The chemistry and catalysis of the water gas shift reaction: 1. The kinetics over supported metal catalysts. J. Catal. 1981, 67, 90–102. [Google Scholar] [CrossRef]
- Grilc, M.; Likozar, B.; Levec, J. Hydrodeoxygenation and hydrocracking of solvolysed lignocellulosic biomass by oxide, reduced and sulphide form of NiMo, Ni, Mo and Pd catalysts. Appl. Catal. B Environ. 2014, 150, 275–287. [Google Scholar] [CrossRef]
- Taylor, W.A.; Acree, S.F. The Equilibrium between Mucic Acid and Its Lactones. J. Phys. Chem. 1915, 20, 118–120. [Google Scholar] [CrossRef]
- Brown, J.M.; Manley-Harris, M.; Field, R.J.; Kiely, D.E. An NMR Study of the Equilibration of d-Glucaric Acid with Lactone Forms in Aqueous Acid Solutions. J. Carbohydr. Chem. 2007, 26, 455–467. [Google Scholar] [CrossRef]
- De Jong, E.; Dam, M.A.; Sipos, L.; Gruter, G.-J.M. Furandicarboxylic Acid (FDCA), A Versatile Building Block for a Very Interesting Class of Polyesters. Biobased Monomers Polym. Mater. 2012, 105, 1–13. [Google Scholar]
- Marcus, R.; Regina, P. Cellulose-Based Sustainable Polymers: State of the Art and Future Trends. Macromol. Rapid Commun. 2011, 32, 1299–1311. [Google Scholar]
- Unnikrishnan, P.; Srinivas, D. Chapter 3-Heterogeneous Catalysis A2-Joshi, S. In Industrial Catalytic Processes for Fine and Specialty Chemicals; Ranade, V.V., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 41–111. ISBN 978-0-12-801457-8. [Google Scholar]
- Watson, M.J. Platinum Group Metal Catalysed Hydrodeoxygenation Of Model Bio-oil Compounds. Johnson Matthey Technol. Rev. 2014, 58, 156–161. [Google Scholar] [CrossRef]
- Lee, H.; Kim, H.; Yu, M.J.; Ko, C.H.; Jeon, J.-K.; Jae, J.; Park, S.H.; Jung, S.-C.; Park, Y.-K. Catalytic Hydrodeoxygenation of Bio-oil Model Compounds over Pt/HY Catalyst. Sci. Rep. 2016, 6, 28765. [Google Scholar] [CrossRef]
- Levene, P.A.; Simms, H.S. Lactone Formation from Mono- and Dicarboxylic Sugar Acids. J. Biol. Chem. 1925, 65, 31–47. [Google Scholar]
- Trueba, M.; Trasatti, S.P. γ-Alumina as a Support for Catalysts: A Review of Fundamental Aspects. Eur. J. Inorg. Chem. 2005, 17, 3393–3403. [Google Scholar] [CrossRef]
- Leofanti, G.; Padovan, M.; Tozzola, G.; Venturelli, B. Surface area and pore texture of catalysts. Catal. Today 1998, 41, 207–219. [Google Scholar] [CrossRef]
- Trimm, D.L.; Stanislaus, A. The control of pore size in alumina catalyst supports: A review. Appl. Catal. 1986, 21, 215–238. [Google Scholar] [CrossRef]
- He, Z.; Wang, X. Hydrodeoxygenation of model compounds and catalytic systems for pyrolysis bio-oils upgrading. Catal. Sustain. Energy 2012, 1, 28. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Y.; Zheng, H.; Zhang, C.; Zhang, B.; Li, Y. Aqueous-phase hydrodeoxygenation of carboxylic acids to alcohols or alkanes over supported Ru catalysts. J. Mol. Catal. A Chem. 2011, 351, 217–227. [Google Scholar] [CrossRef]
- Ruddy, D.A.; Schaidle, J.A.; Ferrell Iii, J.R.; Wang, J.; Moens, L.; Hensley, J.E. Recent advances in heterogeneous catalysts for bio-oil upgrading via “ex situ catalytic fast pyrolysis”: Catalyst development through the study of model compounds. Green Chem. 2014, 16, 454–490. [Google Scholar] [CrossRef]
- Xiong, H.; Pham, H.N.; Datye, A.K. Hydrothermally stable heterogeneous catalysts for conversion of biorenewables. Green Chem. 2014, 16, 4627–4643. [Google Scholar] [CrossRef]
- Van Cleve, T.; Underhill, D.; Veiga Rodrigues, M.; Sievers, C.; Medlin, J.W. Enhanced Hydrothermal Stability of γ-Al2O3 Catalyst Supports with Alkyl Phosphonate Coatings. Langmuir 2018, 34, 3619–3625. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
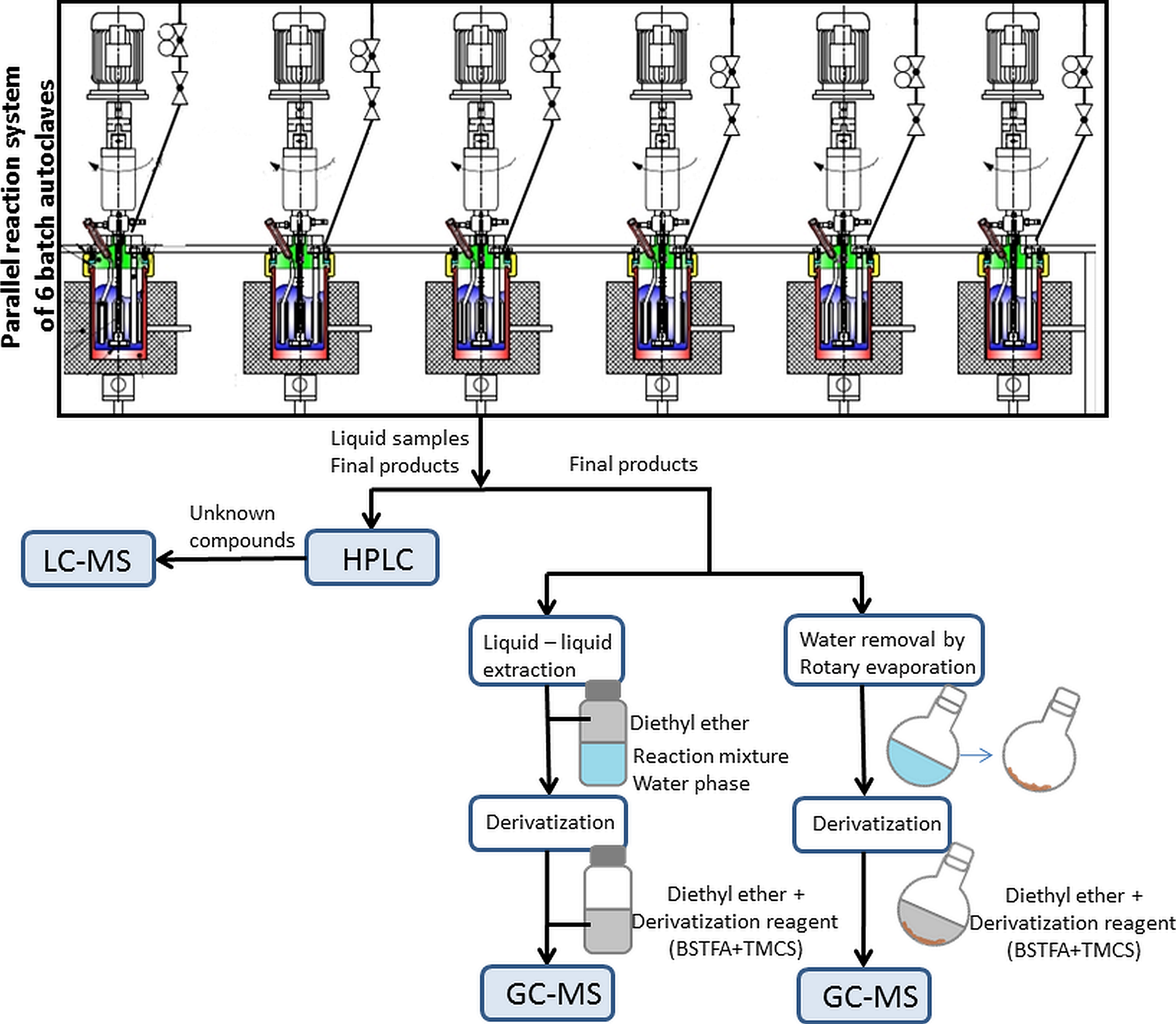{kind=link}
| Catalyst | Metal Loading (wt%) | SBET (m2 g−1) | Vp (cm3 g−1) | Dp (nm) |
|---|---|---|---|---|
| NiMo/γ-Al2O3 | 3/15 1 | 123 | 0.29 | 9.6 |
| Pt/γ-Al2O3 | 5 | 97 | 0.23 | 9.5 |
| Rh/SiO2 | 5 | 147 | 0.58 | 15.5 |
| Pt/SiO2 | 5 | 144 | 0.67 | 18.7 |
| Ni/SiO2 | 5 | 137 | 0.58 | 16.6 |
| Ru/SiO2 | 5 | 154 | 0.66 | 17.1 |
| Ru/C | 5 | 745 | 0.74 | 4.0 |
| Ni/C | 5 | 1017 | 0.57 | 2.6 |
| Pt/C | 5 | 1275 | 1.22 | 3.7 |
| Rh/C | 5 | 1032 | 0.58 | 2.3 |
| Prod No. | Name of Compound | Molecular Formula | Molar Mass (g mol−1) | Ret. Time (HPLC) (min) | Ret. Time (GC-MS) (min) |
|---|---|---|---|---|---|
| 1 | Mucic acid | C6H10O8 | 210 | 2.3 | - |
| 2 | Mucic-1,4-lactoneMucic-3,6-lactone | C6H8O7 C6H8O7 | 192 192 | 2.7 2.6 | - |
| 3 | Galacturonic acid | C6H10O7 | 194 | 2.1 | - |
| 4 | Galactonic acid | C6H12O7 | 196 | 1.9 | - |
| 5 | 2,5-dihydroxy dihexenoic acid | C6H8O6 | 176 | - | - |
| 6 | 2,3-dihydroxy dihexenoic acid | C6H8O6 | 176 | - | - |
| 7 | 2,5-dihydroxy-6-oxo-hexenoic acid | C6H8O5 | 160 | - | - |
| 8 | Furandicarboxylic acid | C6H4O5 | 156 | 9.5 | - |
| 9 | Muconic acid | C6H6O4 | 142 | 10.3 | - |
| 10 | Adipic acid | C6H10O4 | 146 | 9.9 | 18.4 |
| 11 | 2-hydroxy hexanoic acid | C6H10O5 | 162 | 11.3 | 17.2 |
| 12 | Hexanoic acid | C6H12O2 | 116 | 12.7 | 15.7 |
| 13 | 1,6-Hexanediol | C6H14O2 | 118 | - | - |
| 14 | Hexane | C6H14 | 86 | - | - |
| 15 | 1,4-dihydroxy pentenoic acid | C5H8O4 | 132 | - | - |
| 16 | 2-furoic acid | C5H4O3 | 112 | - | 15.5 |
| 17 | 2,5-Dihydro-2-furancarboxylic acid | C5H6O3 | 114 | - | - |
| 18 | Tetrahydro-2-furancarboxylic acid | C5H8O3 | 116 | 6.3 | - |
| 19 | Tetrahydro-2-furfuryl alcohol | C5H10O2 | 102 | 11.3 | 15.6 |
| 20 | 2-Pentenoic acid | C5H8O2 | 100 | - | 13.8 |
| 21 | Pentanoic acid | C5H10O2 | 102 | - | 14.5 |
| 22 | Pentanal | C5H10O | 86 | - | - |
| 23 | 1-Pentanol | C5H12O | 88 | - | - |
| 24 | Pentane | C5H12 | 72 | - | - |
| 25 | 1,2-dihydroxy pentenoic acid | C5H8O4 | 132 | - | - |
| 26 | Levulinic acid | C5H8O3 | 116 | 5.6 | 15.45 |
| 27 | 2-hydroxy pentanoic acid | C5H10O3 | 118 | - | 15.91 |
| 28 | 4-hydroxy pentenoic acid | C5H10O3 | 118 | - | 15.84 |
| 29 | Butane | C4H10 | 58 | - | - |
| 30 | Butyric acid | C4H8O2 | 88 | - | 12.77 |
| 31 | Lactic acid | C3H6O3 | 90 | 3.3 | 12.85 |
| Name | Molar Fraction in the Diethyl Ether Extract 1 | Yields in the Reaction Mixture 2 | ||
|---|---|---|---|---|
| T = 200 °C (mol%) | T = 225 °C (mol%) | T = 200 °C (mol%) | T = 225 °C (mol%) | |
| Tetrahydro-2-furfuryl alcohol | 36.0 | 12.4 | 10.1 | - |
| Adipic acid | 24.3 | 32.3 | 4.25 | 4.33 |
| 2-Hydroxypentanoic acid | 7.4 | 5.6 | - | - |
| Levulinic acid | 5.5 | 5.0 | 1.12 | 3.08 |
| 2-Furoic acid | 6.3 | 25.4 | 0.14 | 0.39 |
| 2-Pentenoic acid | 3.6 | 1.8 | - | - |
| 2-Hydroxyhexanoic acid | 0.8 | 2.1 | 1.48 | 1.08 |
| 4-Pentenoic acid | 3.2 | 2.4 | - | - |
| 2-Methylpropanoic acid | 2.2 | - | - | - |
| Hexanoic acid | 1.6 | 1.3 | - | - |
| 2-Methyl-1-butanol | 1.0 | 1.0 | - | - |
| Butyric acid | 2.8 | 2.8 | - | - |
| Propanoic acid | 0.9 | 1.2 | - | - |
| 2-Butene-1,4-diol | 0.7 | - | - | - |
| 4-Hydroxypentanoic acid | 0.8 | 0.9 | - | - |
| 3-Methyl-2-hydroxypentanoic acid | 3.0 | 5.8 | - | - |
| Experiment Number | Catalyst | T (°C) | PH2 (MPa) | PN2 (MPa) | Reactant/Catalyst Ratio (/) |
|---|---|---|---|---|---|
| 1 | NiMo/γ-Al2O3 | 125 | 5 | 0 | 0.4 |
| 2 | NiMo/γ-Al2O3 | 135 | 5 | 0 | 0.4 |
| 3 | NiMo/γ-Al2O3 | 150 | 5 | 0 | 0.4 |
| 4 | NiMo/γ-Al2O3 | 175 | 5 | 0 | 0.4 |
| 5 | NiMo/γ-Al2O3 | 200 | 5 | 0 | 0.4 |
| 6 | NiMo/γ-Al2O3 | 225 | 5 | 0 | 0.4 |
| 7 | Pt/γ-Al2O3 | 125 | 5 | 0 | 0.4 |
| 8 | Pt/γ-Al2O3 | 135 | 5 | 0 | 0.4 |
| 9 | Pt/γ-Al2O3 | 150 | 5 | 0 | 0.4 |
| 10 | Pt/γ-Al2O3 | 175 | 5 | 0 | 0.4 |
| 11 | Rh/SiO2 | 125 | 5 | 0 | 0.4 |
| 12 | Rh/SiO2 | 135 | 5 | 0 | 0.4 |
| 13 | Rh/SiO2 | 150 | 5 | 0 | 0.4 |
| 14 | Rh/SiO2 | 175 | 5 | 0 | 0.4 |
| 15 | Pt/SiO2 | 125 | 5 | 0 | 0.4 |
| 16 | Pt/SiO2 | 135 | 5 | 0 | 0.4 |
| 17 | Pt/SiO2 | 150 | 5 | 0 | 0.4 |
| 18 | Pt/SiO2 | 175 | 5 | 0 | 0.4 |
| 19 | Ni/SiO2 | 125 | 5 | 0 | 0.4 |
| 20 | Ni/SiO2 | 135 | 5 | 0 | 0.4 |
| 21 | Ni/SiO2 | 150 | 5 | 0 | 0.4 |
| 22 | Ni/SiO2 | 175 | 5 | 0 | 0.4 |
| 23 | Ru/SiO2 | 125 | 5 | 0 | 0.4 |
| 24 | Ru/SiO2 | 135 | 5 | 0 | 0.4 |
| 25 | Ru/SiO2 | 150 | 5 | 0 | 0.4 |
| 26 | Ru/SiO2 | 175 | 5 | 0 | 0.4 |
| 27 | Ru/C | 125 | 5 | 0 | 0.4 |
| 28 | Ru/C | 135 | 5 | 0 | 0.4 |
| 29 | Ru/C | 150 | 5 | 0 | 0.4 |
| 30 | Ru/C | 175 | 5 | 0 | 0.4 |
| 31 | Ni/C | 125 | 5 | 0 | 0.4 |
| 32 | Ni/C | 135 | 5 | 0 | 0.4 |
| 33 | Ni/C | 150 | 5 | 0 | 0.4 |
| 34 | Ni/C | 175 | 5 | 0 | 0.4 |
| 35 | Pt/C | 125 | 5 | 0 | 0.4 |
| 36 | Pt/C | 135 | 5 | 0 | 0.4 |
| 37 | Pt/C | 150 | 5 | 0 | 0.4 |
| 38 | Pt/C | 175 | 5 | 0 | 0.4 |
| 39 | Rh/C | 125 | 5 | 0 | 0.4 |
| 40 | Rh/C | 135 | 5 | 0 | 0.4 |
| 41 | Rh/C | 150 | 5 | 0 | 0.4 |
| 42 | Rh/C | 175 | 5 | 0 | 0.4 |
| 43 | No cat. | 125 | 5 | 0 | - |
| 44 | No cat. | 135 | 5 | 0 | - |
| 45 | No cat. | 150 | 5 | 0 | - |
| 46 | No cat. | 175 | 5 | 0 | - |
| 47 | No cat. | 125 | 0 | 5 | - |
| 48 | No cat. | 135 | 0 | 5 | - |
| 49 | No cat. | 150 | 0 | 5 | - |
| 50 | No cat. | 175 | 0 | 5 | - |
| 51 | Carbon | 150 | 5 | 0 | 0.4 |
| 52 | Silica | 150 | 5 | 0 | 0.4 |
| 53 | Alumina | 150 | 5 | 0 | 0.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hočevar, B.; Grilc, M.; Likozar, B. Aqueous Dehydration, Hydrogenation, and Hydrodeoxygenation Reactions of Bio-Based Mucic Acid over Ni, NiMo, Pt, Rh, and Ru on Neutral or Acidic Catalyst Supports. Catalysts 2019, 9, 286. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9030286
Hočevar B, Grilc M, Likozar B. Aqueous Dehydration, Hydrogenation, and Hydrodeoxygenation Reactions of Bio-Based Mucic Acid over Ni, NiMo, Pt, Rh, and Ru on Neutral or Acidic Catalyst Supports. Catalysts. 2019; 9(3):286. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9030286
Chicago/Turabian StyleHočevar, Brigita, Miha Grilc, and Blaž Likozar. 2019. "Aqueous Dehydration, Hydrogenation, and Hydrodeoxygenation Reactions of Bio-Based Mucic Acid over Ni, NiMo, Pt, Rh, and Ru on Neutral or Acidic Catalyst Supports" Catalysts 9, no. 3: 286. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9030286