Arachidonic Acid Evokes an Increase in Intracellular Ca2+ Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Solutions
2.3. [Ca2+]i and NO Imaging
2.4. Western Blotting
2.5. Statistical Analysis
2.6. Chemicals
3. Results
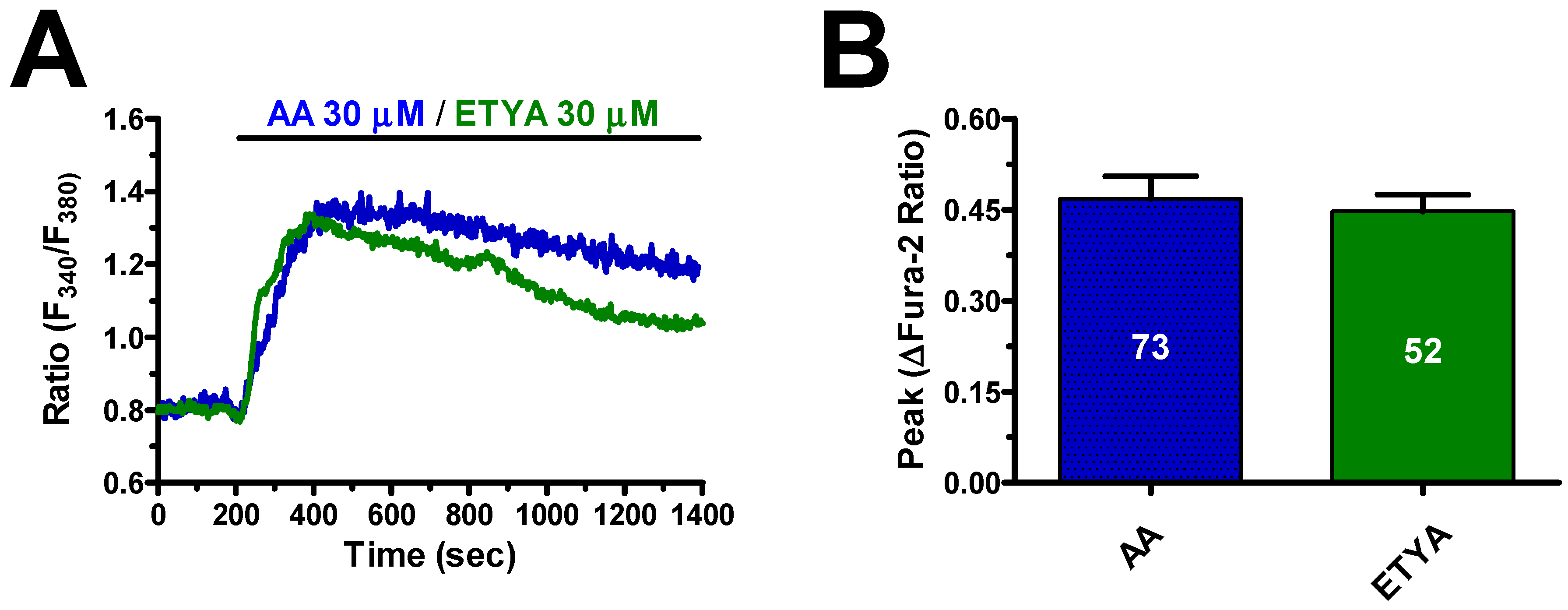
3.1. AA Triggers an Increase in [Ca2+]i in hCMEC/D3 Cells
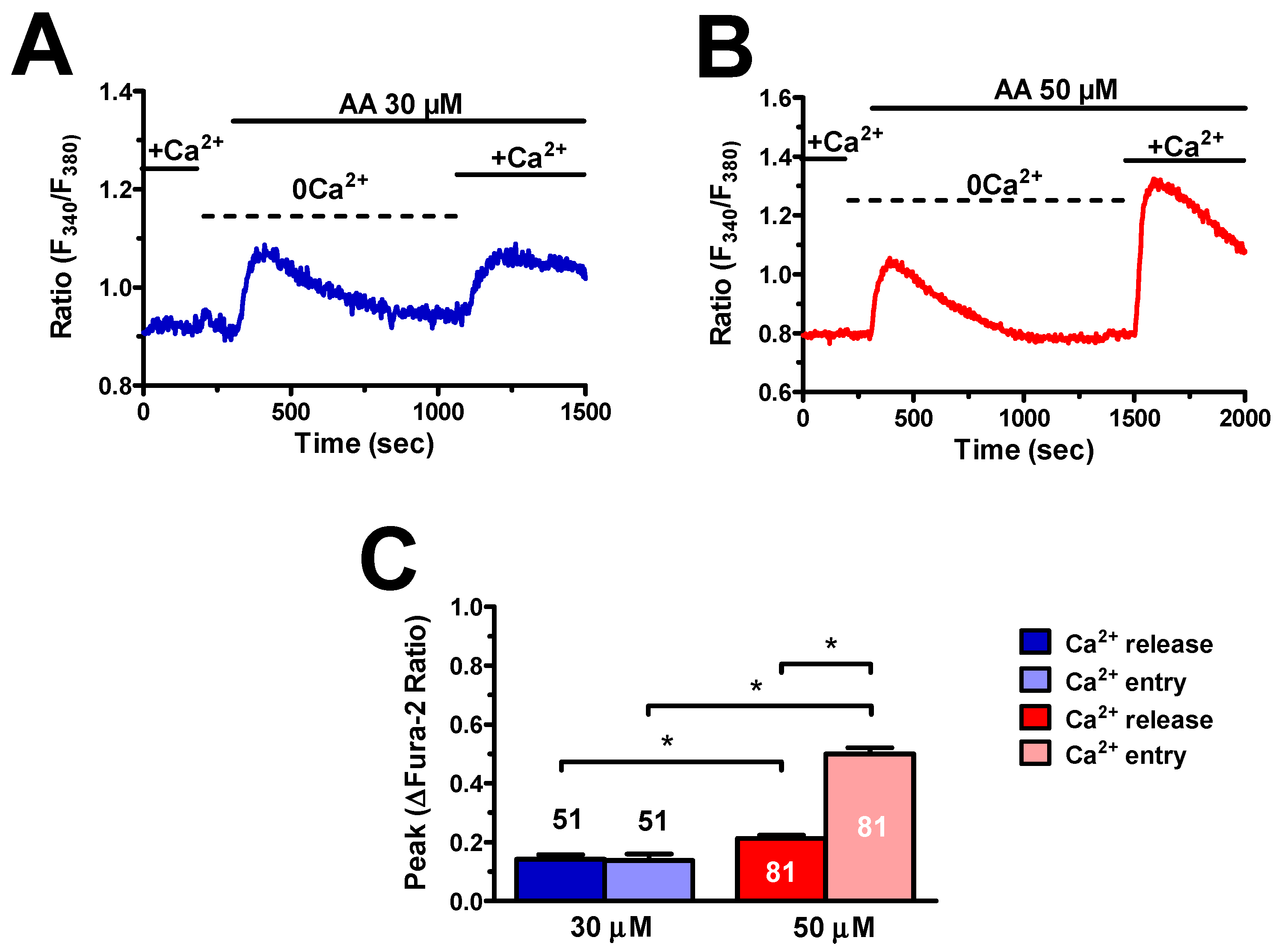
3.2. AA-Induced Intracellular Ca2+ Signals Required Endogenous Ca2+ Release and Extracellular Ca2+ Influx
3.3. InsP3R3 and TPC1-2 Mediate AA-Induced Endogenous Ca2+ Release
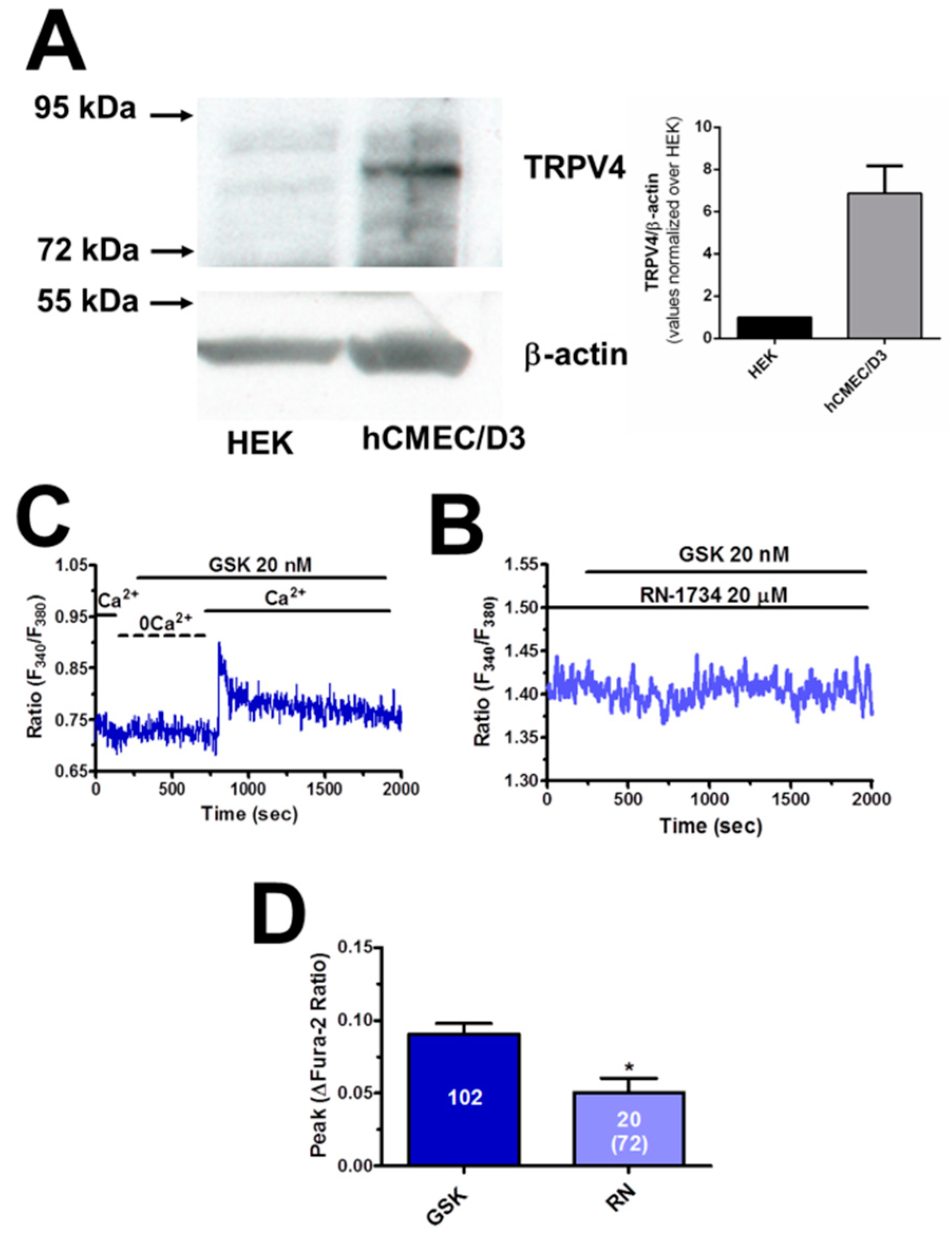
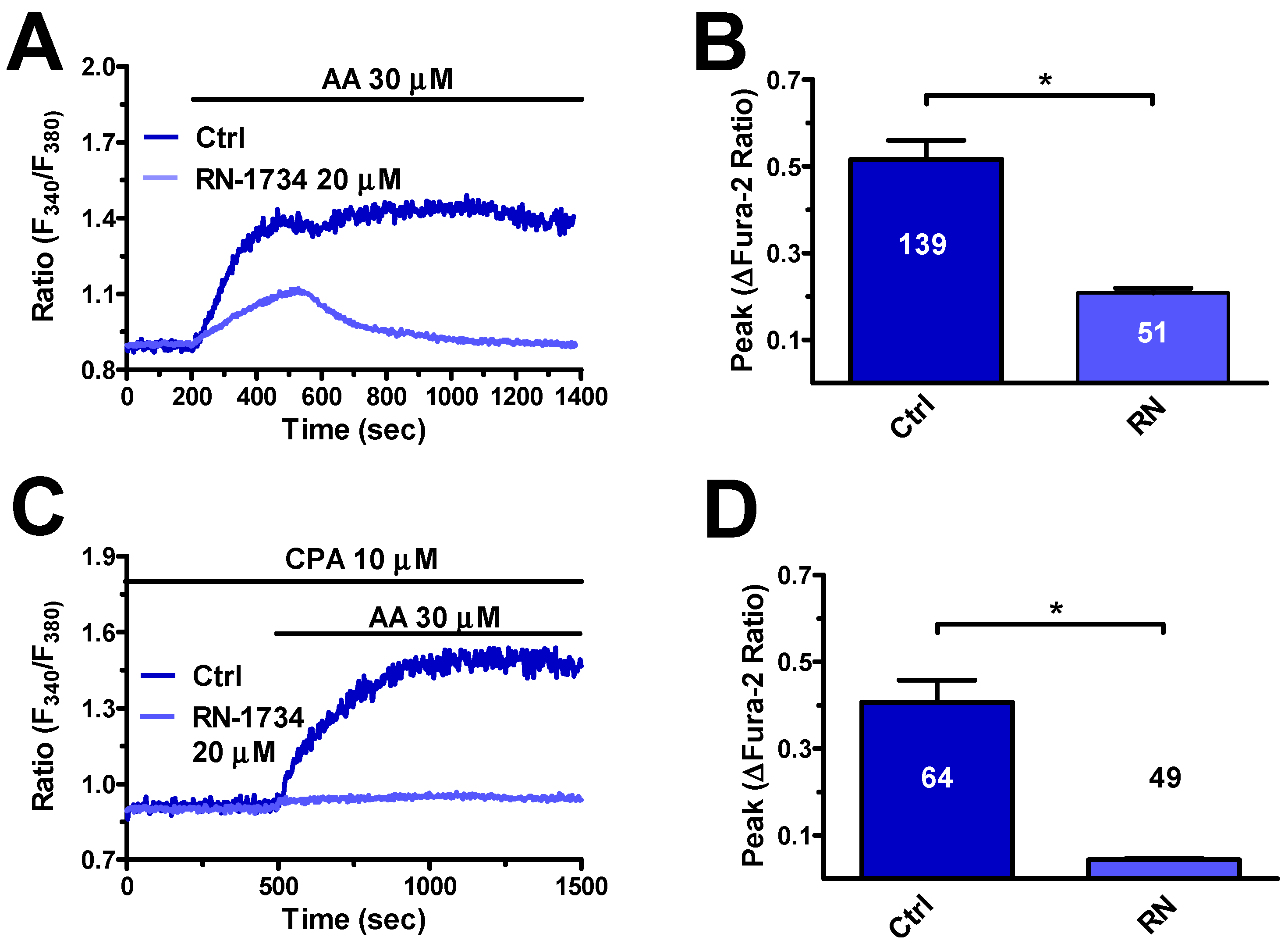
3.4. TRPV4 is Expressed and Mediates AA-Evoked Extracellular Ca2+ Influx
3.5. AA-Evoked Intracellular Ca2+ Signaling Drives NO Release in hCMEC/D3 Cells
4. Discussion
4.1. Intracellular Ca2+ Signals Induced by AA in hCMEC/D3 Cells
4.2. The Dose–Response Relationship of AA-Evoked Intracellular Ca2+ Signals in hCMEC/D3 Cells
4.3. The Complex Mechanism of AA-Evoked Intracellular Ca2+ Signals in hCMEC/D3 Cells: InsP3R3, TPC1-2, and TRPV4
4.3.1. InsP3R3 and TPC1-2
4.3.2. TRPV4
4.4. AA Induces NO Release in hCMEC/D3 Cells: The Rationale for the Present Investigation
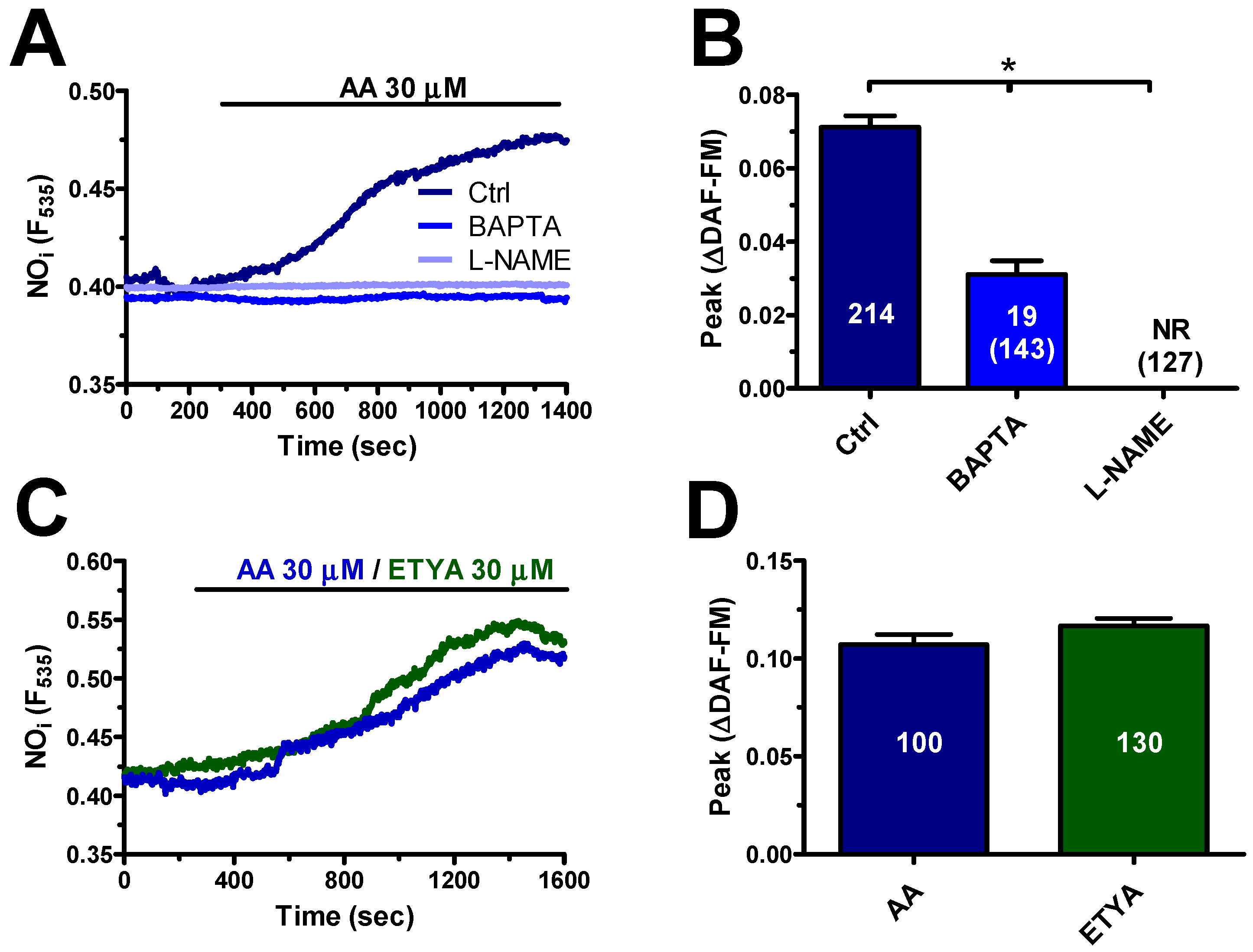
4.4.1. AA Induces Delayed NO Release in hCMEC/D3 Cells
4.4.2. AA Induced NO Release via TPC1-2 and TRPV4 in hCMEC/D3 Cells
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Munaron, L. Shuffling the cards in signal transduction: Calcium, arachidonic acid and mechanosensitivity. World J. Biol. Chem. 2011, 2, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Harder, D.R.; Rarick, K.R.; Gebremedhin, D.; Cohen, S.S. Regulation of Cerebral Blood Flow: Response to Cytochrome P450 Lipid Metabolites. Compr. Physiol. 2018, 8, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Mapelli, L.; Gagliano, G.; Soda, T.; Laforenza, U.; Moccia, F.; D’Angelo, E.U. Granular Layer Neurons Control Cerebellar Neurovascular Coupling Through an NMDA Receptor/NO-Dependent System. J. Neurosci. 2017, 37, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, B.A.; Newman, E.A. Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Lecrux, C.; Hamel, E. Neuronal networks and mediators of cortical neurovascular coupling responses in normal and altered brain states. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef] [PubMed]
- Lecrux, C.; Toussay, X.; Kocharyan, A.; Fernandes, P.; Neupane, S.; Levesque, M.; Plaisier, F.; Shmuel, A.; Cauli, B.; Hamel, E. Pyramidal neurons are “neurogenic hubs” in the neurovascular coupling response to whisker stimulation. J. Neurosci. 2011, 31, 9836–9847. [Google Scholar] [CrossRef] [PubMed]
- Lecrux, C.; Kocharyan, A.; Sandoe, C.H.; Tong, X.K.; Hamel, E. Pyramidal cells and cytochrome P450 epoxygenase products in the neurovascular coupling response to basal forebrain cholinergic input. J. Cereb. Blood Flow Metab. 2012, 32, 896–906. [Google Scholar] [CrossRef]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Hogan-Cann, A.D.; Lu, P.; Anderson, C.M. Endothelial NMDA receptors mediate activity-dependent brain hemodynamic responses in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 10229–10231. [Google Scholar] [CrossRef] [Green Version]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca(2+) Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca(2+) oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca(2+) release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef] [PubMed]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca(2+) signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef]
- Mottola, A.; Antoniotti, S.; Lovisolo, D.; Munaron, L. Regulation of noncapacitative calcium entry by arachidonic acid and nitric oxide in endothelial cells. Faseb J. 2005, 19, 2075–2077. [Google Scholar] [CrossRef] [Green Version]
- Fiorio Pla, A.; Genova, T.; Pupo, E.; Tomatis, C.; Genazzani, A.; Zaninetti, R.; Munaron, L. Multiple roles of protein kinase a in arachidonic acid-mediated Ca2+ entry and tumor-derived human endothelial cell migration. Mol. Cancer Res. 2010, 8, 1466–1476. [Google Scholar] [CrossRef]
- Kohler, R.; Heyken, W.T.; Heinau, P.; Schubert, R.; Si, H.; Kacik, M.; Busch, C.; Grgic, I.; Maier, T.; Hoyer, J. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arter. Thromb. Vasc. Biol. 2006, 26, 1495–1502. [Google Scholar] [CrossRef]
- Fiorio Pla, A.; Munaron, L. Calcium influx, arachidonic acid, and control of endothelial cell proliferation. Cell Calcium 2001, 30, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vascul. Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.; Ko, Y.; Mata, W.; Saquib, M.; Eldridge, J.; Cohen-Gadol, A.; Leaver, H.A.; Wang, S.; Rizzo, M.T. Arachidonic acid induces brain endothelial cell apoptosis via p38-MAPK and intracellular calcium signaling. Microvasc. Res. 2015, 98, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Peppiatt, C.M.; Holmes, A.M.; Seo, J.T.; Bootman, M.D.; Collins, T.J.; McDonald, F.; Roderick, H.L. Calmidazolium and arachidonate activate a calcium entry pathway that is distinct from store-operated calcium influx in HeLa cells. Biochem. J. 2004, 381, 929–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, A.M.; Roderick, H.L.; McDonald, F.; Bootman, M.D. Interaction between store-operated and arachidonate-activated calcium entry. Cell Calcium 2007, 41, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turovsky, E.A.; Zinchenko, V.P.; Kaimachnikov, N.P. Attenuation of calmodulin regulation evokes Ca(2+) oscillations: Evidence for the involvement of intracellular arachidonate-activated channels and connexons. Mol. Cell. Biochem. 2019, 456, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wu, J.; Hu, M.; Wu, J.; Zhu, Q.; Yang, Z.; Xie, X.; Feng, Y.Q.; Yue, J. Glutamate affects the CYP1B1- and CYP2U1-mediated hydroxylation of arachidonic acid metabolism via astrocytic mGlu5 receptor. Int. J. Biochem. Cell Biol. 2019, 110, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Rietdorf, K.; Collins, T.; Walker, S.; Sanderson, M. Ca2+-sensitive fluorescent dyes and intracellular Ca2+ imaging. Cold Spring Harb. Protoc. 2013, 2013, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.H.; Errington, M.L.; Lynch, M.A.; Bliss, T.V. Arachidonic acid induces a long-term activity-dependent enhancement of synaptic transmission in the hippocampus. Nature 1989, 341, 739–742. [Google Scholar] [CrossRef]
- Kato, K.; Uruno, K.; Saito, K.; Kato, H. Both arachidonic acid and 1-oleoyl-2-acetyl glycerol in low magnesium solution induce long-term potentiation in hippocampal CA1 neurons in vitro. Brain Res. 1991, 563, 94–100. [Google Scholar] [CrossRef]
- Bazan, N.G. Synaptic lipid signaling: Significance of polyunsaturated fatty acids and platelet-activating factor. J. Lipid Res. 2003, 44, 2221–2233. [Google Scholar] [CrossRef]
- Watanabe, H.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T.; Nilius, B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 2003, 424, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Fisslthaler, B.; Suzuki, M.; Janssens, A.; Voets, T.; Morisseau, C.; Hammock, B.D.; Fleming, I.; Busse, R.; et al. Modulation of the Ca2+ permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ. Res. 2005, 97, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Loot, A.E.; Popp, R.; Fisslthaler, B.; Vriens, J.; Nilius, B.; Fleming, I. Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc. Res. 2008, 80, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, M.Y.; King, J.A.; Al-Mehdi, A.B.; Liedtke, W.; Townsley, M.I. High vascular pressure-induced lung injury requires P450 epoxygenase-dependent activation of TRPV4. Am. J. Respir Cell Mol. Biol. 2008, 38, 386–392. [Google Scholar] [CrossRef]
- Li, J.; Bruns, A.F.; Hou, B.; Rode, B.; Webster, P.J.; Bailey, M.A.; Appleby, H.L.; Moss, N.K.; Ritchie, J.E.; Yuldasheva, N.Y.; et al. Orai3 Surface Accumulation and Calcium Entry Evoked by Vascular Endothelial Growth Factor. Arter. Thromb Vasc. Biol. 2015, 35, 1987–1994. [Google Scholar] [CrossRef] [Green Version]
- Fiorio Pla, A.; Grange, C.; Antoniotti, S.; Tomatis, C.; Merlino, A.; Bussolati, B.; Munaron, L. Arachidonic acid-induced Ca2+ entry is involved in early steps of tumor angiogenesis. Mol. Cancer Res. 2008, 6, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Yeung-Yam-Wah, V.; Lee, A.K.; Tse, A. Arachidonic acid mobilizes Ca2+ from the endoplasmic reticulum and an acidic store in rat pancreatic β cells. Cell Calcium 2012, 51, 140–148. [Google Scholar] [CrossRef]
- Moccia, F.; Dragoni, S.; Poletto, V.; Rosti, V.; Tanzi, F.; Ganini, C.; Porta, C. Orai1 and Transient Receptor Potential Channels as novel molecular targets to impair tumor neovascularisation in renal cell carcinoma and other malignancies. Anticancer Agents Med. Chem. 2014, 14, 296–312. [Google Scholar] [CrossRef]
- Pitt, S.J.; Reilly-O’Donnell, B.; Sitsapesan, R. Exploring the biophysical evidence that mammalian two-pore channels are NAADP-activated calcium-permeable channels. J. Physiol. 2016, 594, 4171–4179. [Google Scholar] [CrossRef] [Green Version]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca(2+) Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers 2018, 11, 27. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Schapira, A.H.; Futter, C.E.; Patel, S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci. 2013, 126, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Ong, H.L.; Cheng, K.T.; Brossa, A.; Bussolati, B.; Lockwich, T.; Paria, B.; Munaron, L.; Ambudkar, I.S. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 2012, 31, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Remodelling of the Ca2+ Toolkit in Tumor Endothelium as a Crucial Responsible for the Resistance to Anticancer Therapies. Curr. Signal. Transd. Ther. 2017, 12. [Google Scholar] [CrossRef]
- Dragoni, S.; Guerra, G.; Fiorio Pla, A.; Bertoni, G.; Rappa, A.; Poletto, V.; Bottino, C.; Aronica, A.; Lodola, F.; Cinelli, M.P.; et al. A functional Transient Receptor Potential Vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J. Cell. Physiol. 2015, 230, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Thoppil, R.J.; Adapala, R.K.; Cappelli, H.C.; Kondeti, V.; Dudley, A.C.; Gary Meszaros, J.; Paruchuri, S.; Thodeti, C.K. TRPV4 channel activation selectively inhibits tumor endothelial cell proliferation. Sci. Rep. 2015, 5, 14257. [Google Scholar] [CrossRef]
- White, J.P.; Cibelli, M.; Urban, L.; Nilius, B.; McGeown, J.G.; Nagy, I. TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol. Rev. 2016, 96, 911–973. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef]
- Zuccolo, E.; Bottino, C.; Diofano, F.; Poletto, V.; Codazzi, A.C.; Mannarino, S.; Campanelli, R.; Fois, G.; Marseglia, G.L.; Guerra, G.; et al. Constitutive Store-Operated Ca(2+) Entry Leads to Enhanced Nitric Oxide Production and Proliferation in Infantile Hemangioma-Derived Endothelial Colony-Forming Cells. Stem Cells Dev. 2016, 25, 301–319. [Google Scholar] [CrossRef]
- Carta, M.; Lanore, F.; Rebola, N.; Szabo, Z.; Da Silva, S.V.; Lourenco, J.; Verraes, A.; Nadler, A.; Schultz, C.; Blanchet, C.; et al. Membrane lipids tune synaptic transmission by direct modulation of presynaptic potassium channels. Neuron 2014, 81, 787–799. [Google Scholar] [CrossRef]
- Harraz, O.F.; Longden, T.A.; Hill-Eubanks, D.; Nelson, M.T. PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Fleming, N.; Mellow, L. Arachidonic acid stimulates intracellular calcium mobilization and regulates protein synthesis, ATP levels, and mucin secretion in submandibular gland cells. J. Dent. Res. 1995, 74, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Rimessi, A.; Giorgi, C.; Baldini, C.; Ferroni, L.; Rizzuto, R.; Pinton, P. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem. Biophys. Res. Commun. 2008, 375, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Marchi, S.; Bonora, M.; Aguiari, P.; Bononi, A.; De Stefani, D.; Giorgi, C.; Leo, S.; Rimessi, A.; Siviero, R.; et al. Ca(2+) transfer from the ER to mitochondria: When, how and why. Biochim. Biophys. Acta 2009, 1787, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Pellavio, G.; Ferulli, F.; Di Nezza, F.; Shekha, M.; Lim, D.; Maestri, M.; Guerra, G.; Ambrosone, L.; Pedrazzoli, P.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca(2+) Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells. Cancers 2019, 11, 542. [Google Scholar] [CrossRef] [PubMed]
- Ronco, V.; Potenza, D.M.; Denti, F.; Vullo, S.; Gagliano, G.; Tognolina, M.; Guerra, G.; Pinton, P.; Genazzani, A.A.; Mapelli, L.; et al. A novel Ca(2)(+)-mediated cross-talk between endoplasmic reticulum and acidic organelles: Implications for NAADP-dependent Ca(2)(+) signalling. Cell Calcium 2015, 57, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5, e15887. [Google Scholar] [CrossRef]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca(2+) to Lysosomes. Cell Rep. 2018, 25, 3180–3193.e7. [Google Scholar] [CrossRef]
- Di Nezza, F.; Zuccolo, E.; Poletto, V.; Rosti, V.; De Luca, A.; Moccia, F.; Guerra, G.; Ambrosone, L. Liposomes as a Putative Tool to Investigate NAADP Signaling in Vasculogenesis. J. Cell. Biochem. 2017, 118, 3722–3729. [Google Scholar] [CrossRef]
- Moccia, F.; Nusco, G.A.; Lim, D.; Kyozuka, K.; Santella, L. NAADP and InsP3 play distinct roles at fertilization in starfish oocytes. Dev. Biol. 2006, 294, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Galione, A. A primer of NAADP-mediated Ca(2+) signalling: From sea urchin eggs to mammalian cells. Cell Calcium 2015, 58, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, X.; Gonzalez-Cobos, J.C.; Stolwijk, J.A.; Matrougui, K.; Trebak, M. Leukotriene-C4 synthase, a critical enzyme in the activation of store-independent Orai1/Orai3 channels, is required for neointimal hyperplasia. J. Biol. Chem. 2015, 290, 5015–5027. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nippert, A.R.; Biesecker, K.R.; Newman, E.A. Mechanisms Mediating Functional Hyperemia in the Brain. Neuroscientist 2018, 24, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Munaron, L. Intracellular calcium, endothelial cells and angiogenesis. Recent Pat. Anticancer Drug Discov. 2006, 1, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Endothelial Ca(2+) Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef]
- Hall, C.N.; Garthwaite, J. What is the real physiological NO concentration in vivo? Nitric Oxide 2009, 21, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Cauli, B.; Hamel, E. Revisiting the role of neurons in neurovascular coupling. Front. Neuroenergetics 2010, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Anfuso, C.D.; Giurdanella, G.; Motta, C.; Muriana, S.; Lupo, G.; Ragusa, N.; Alberghina, M. PKCalpha-MAPK/ERK-phospholipase A2 signaling is required for human melanoma-enhanced brain endothelial cell proliferation and motility. Microvasc. Res. 2009, 78, 338–357. [Google Scholar] [CrossRef]
- Giurdanella, G.; Motta, C.; Muriana, S.; Arena, V.; Anfuso, C.D.; Lupo, G.; Alberghina, M. Cytosolic and calcium-independent phospholipase A(2) mediate glioma-enhanced proangiogenic activity of brain endothelial cells. Microvasc. Res. 2011, 81, 1–17. [Google Scholar] [CrossRef]
- Brailoiu, G.C.; Gurzu, B.; Gao, X.; Parkesh, R.; Aley, P.K.; Trifa, D.I.; Galione, A.; Dun, N.J.; Madesh, M.; Patel, S.; et al. Acidic NAADP-sensitive calcium stores in the endothelium: Agonist-specific recruitment and role in regulating blood pressure. J. Biol. Chem. 2010, 285, 37133–37137. [Google Scholar] [CrossRef] [PubMed]
- Tallini, Y.N.; Brekke, J.F.; Shui, B.; Doran, R.; Hwang, S.M.; Nakai, J.; Salama, G.; Segal, S.S.; Kotlikoff, M.I. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: Measurements in Cx40BAC GCaMP2 transgenic mice. Circ. Res. 2007, 101, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.H.T.; Peringod, G.; Gordon, G.R. Astrocytes Integrate Behavioral State and Vascular Signals during Functional Hyperemia. Neuron 2018, 100, 1133–1148.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic Acid Evokes an Increase in Intracellular Ca2+ Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation. Cells 2019, 8, 689. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8070689
Berra-Romani R, Faris P, Negri S, Botta L, Genova T, Moccia F. Arachidonic Acid Evokes an Increase in Intracellular Ca2+ Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation. Cells. 2019; 8(7):689. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8070689
Chicago/Turabian StyleBerra-Romani, Roberto, Pawan Faris, Sharon Negri, Laura Botta, Tullio Genova, and Francesco Moccia. 2019. "Arachidonic Acid Evokes an Increase in Intracellular Ca2+ Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation" Cells 8, no. 7: 689. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8070689