Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis
by
 , , and
, , and
Sharon Negri
1, 
Pawan Faris
1,
Vittorio Rosti
2,
Maria Rosa Antognazza
3,
Francesco Lodola
3 and
Francesco Moccia
1,* 1
Laboratory of General Physiology, Department of Biology and Biotechnology “L. Spallanzani”, University of Pavia, 27100 Pavia, Italy
2
Center for the Study of Myelofibrosis, Laboratory of Biochemistry, Biotechnology and Advanced Diagnosis, IRCCS Policlinico San Matteo Foundation, 27100 Pavia, Italy
3
Center for Nano Science and Technology @PoliMi, Istituto Italiano di Tecnologia, via Pascoli 70/3, 20133 Milano, Italy
*
Author to whom correspondence should be addressed.
Cells 2020, 9(6), 1341; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061341
Submission received: 30 April 2020
/
Revised: 26 May 2020
/
Accepted: 26 May 2020
/
Published: 27 May 2020
(This article belongs to the Special Issue Vascular Signalling)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Therapeutic angiogenesis represents an emerging strategy to treat ischemic diseases by stimulating blood vessel growth to rescue local blood perfusion. Therefore, injured microvasculature may be repaired by stimulating resident endothelial cells or circulating endothelial colony forming cells (ECFCs) or by autologous cell-based therapy. Endothelial Ca2+ signals represent a crucial player in angiogenesis and vasculogenesis; indeed, several angiogenic stimuli induce neovessel formation through an increase in intracellular Ca2+ concentration. Several members of the Transient Receptor Potential (TRP) channel superfamily are expressed and mediate Ca2+-dependent functions in vascular endothelial cells and in ECFCs, the only known truly endothelial precursor. TRP Vanilloid 1 (TRPV1), a polymodal cation channel, is emerging as an important player in endothelial cell migration, proliferation, and tubulogenesis, through the integration of several chemical stimuli. Herein, we first summarize TRPV1 structure and gating mechanisms. Next, we illustrate the physiological roles of TRPV1 in vascular endothelium, focusing our attention on how endothelial TRPV1 promotes angiogenesis. In particular, we describe a recent strategy to stimulate TRPV1-mediated pro-angiogenic activity in ECFCs, in the presence of a photosensitive conjugated polymer. Taken together, these observations suggest that TRPV1 represents a useful target in the treatment of ischemic diseases.
1. Introduction
The interior of blood vessels is lined by endothelial cells, which integrate chemical and physical stimuli deriving from circulating blood and surrounding tissues to redirect blood flow according to local energy requirements [1,2]. Vascular endothelial cells are activated by the local reduction in O2 tension, which follows an ischemic insult to undergo angiogenesis, i.e., the new blood vessel sprouting from adjoining capillaries, thereby attempting to compensate the vascular damage [3]. Additionally, the replacement of injured/apoptotic/senescent vascular endothelial cells may be accomplished by a population of circulating endothelial precursors physically engrafted within neovessels, known as endothelial colony forming cells (ECFCs), which display high proliferative potential and the capability to produce paracrine pro-angiogenic instructions [4,5,6]. Therefore, it has been suggested that ECFCs interact with mature endothelial cells, to maintain vascular integrity and function throughout postnatal life [4,5,6]. While excessive vascularization is associated with tumorigenesis, intraocular and inflammatory diseases, and pulmonary arterial hypertension (PAH) [7,8], insufficient vessel growth or impaired blood flow due to vessel obstruction may lead to severe ischemic disorders, including stroke, peripheral artery disease (PAD), pre-eclampsia, acute myocardial infarction (AMI), and ischemic retinopathies, as well as to neurodegeneration [9,10]. Therapeutic angiogenesis holds great promise for the treatment of ischemic disorders by harnessing multiple pharmacological, cellular, or bioengineering tools to reconstruct the damaged vascular network [11,12]. This strategy postulates that exogenous supplementation of trophic factors, such as growth factors or other bioactive compounds, or transplantation of autologous ECFCs, could foster revascularization in ischemic diseases [5,12,13,14]. Hence, shedding light on the signal transduction pathways driving angiogenesis and vasculogenesis might remarkably impact the regenerative efficiency of therapeutic angiogenesis [5,12,15].
Endothelial Ca2+ signals are known to play an important role in angiogenesis and vasculogenesis. Multiple angiogenic signals (e.g., vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), platelet derived growth factor (PDGF), inflammatory mediators (e.g., thrombin, ATP, ADP, and acetylcholine), pleiotropic hormones (e.g., erythropoietin), and mechanical damage induce endothelial Ca2+ signals, thereby stimulating proliferation, migration, and tube formation, both in vivo and in vitro [16,17,18,19,20,21,22,23,24]. Likewise, VEGF, stromal derived factor-1α (SDF-1α), and the hypoxic human amniotic fluid secretome stimulate distinct intracellular Ca2+ signatures in ECFCs, to promote proliferation, in vitro tubulogenesis, migration, and neovessel formation [25,26,27,28,29]. Vascular endothelial cells and ECFCs rely on endogenous Ca2+ sources, such as endoplasmic reticulum (ER) and endolysosomal vesicles, and on the extracellular environment, to shape the Ca2+ response to pro-angiogenic cues [19,21,30]. Store-operated Ca2+ entry (SOCE), which is recruited upon depletion of the ER Ca2+ reservoir, represents the most common Ca2+ entry pathway upon which inositol triphosphate (InsP3)-synthesizing growth factors and chemokines converge [18,27,30,31,32,33]. An additional endothelial Ca2+ entry route is provided by the superfamily of non-selective Transient Receptor Potential (TRP) cation channels, which comprises 28 members [18,34,35]. Standard nomenclature grouped these 28 members in six subfamilies, based on their sequence homology: canonical (TRPC1-7), vanilloid (TRPV1-6), melastatin (TRPM1-8), ankyrin (TRPA1), mucolipin (TRPML1-3), and polycystin (TRPP). This last subfamily is composed by eight members, although only TRPP2, TRPP3, and TRPP5 display the function and structure of an ion channel [34,36,37]. TRP channels regulate several endothelial functions, mediating extracellular Ca2+ entry and recruiting an array of Ca2+-dependent decoders in response to chemical, thermal, and mechanical stimuli [18,34,35,38]. As recently reviewed in References [18,35], some TRP channel isoforms sustain both angiogenesis and vasculogenesis, including TRPC1 [39,40], TRPC3 [26,41], TRPV4 [42,43], and TRPV1 [42,44]. Interestingly, TRPV1 is rapidly emerging as an important player in endothelial cell migration, proliferation, and tubulogenesis through the integration of distinct environmental and intracellular cues [35,37]. It has recently been proposed that manipulating endothelial Ca2+ signaling could provide an alternative strategy to enhance the vasoreparative outcome of therapeutic angiogenesis and/or to improve the regenerative potential of endogenous endothelial and cardiac progenitors [15,28,45,46]. For instance, it has been suggested that the reparative phenotype of autologous ECFCs could be rejuvenated by transduction with TRPC3 [47], which remarkably increases the pro-angiogenic Ca2+ response to VEGF [26,48]. Based on this rationale, we herein discuss the hypothesis to exploit chemical and physical stimuli to target endothelial TRPV1 for regenerative purposes. First, we summarize current knowledge about the structure, gating mechanisms, and physiological roles of endothelial TRPV1. Then, we illustrate how TRPV1-mediated Ca2+ signals promote angiogenesis in vitro and neovessel formation in vivo in response to a wealth of small molecule agonists that could be harnessed in therapy. Finally, we describe a recent strategy to stimulate TRPV1-dependent pro-angiogenic activity in ECFCs in the presence of a photosensitive organic semiconductor. These evidences hint at TRPV1 as a promising molecular tool to favor post-ischemic revascularization.
2. Vasculogenesis and Angiogenesis
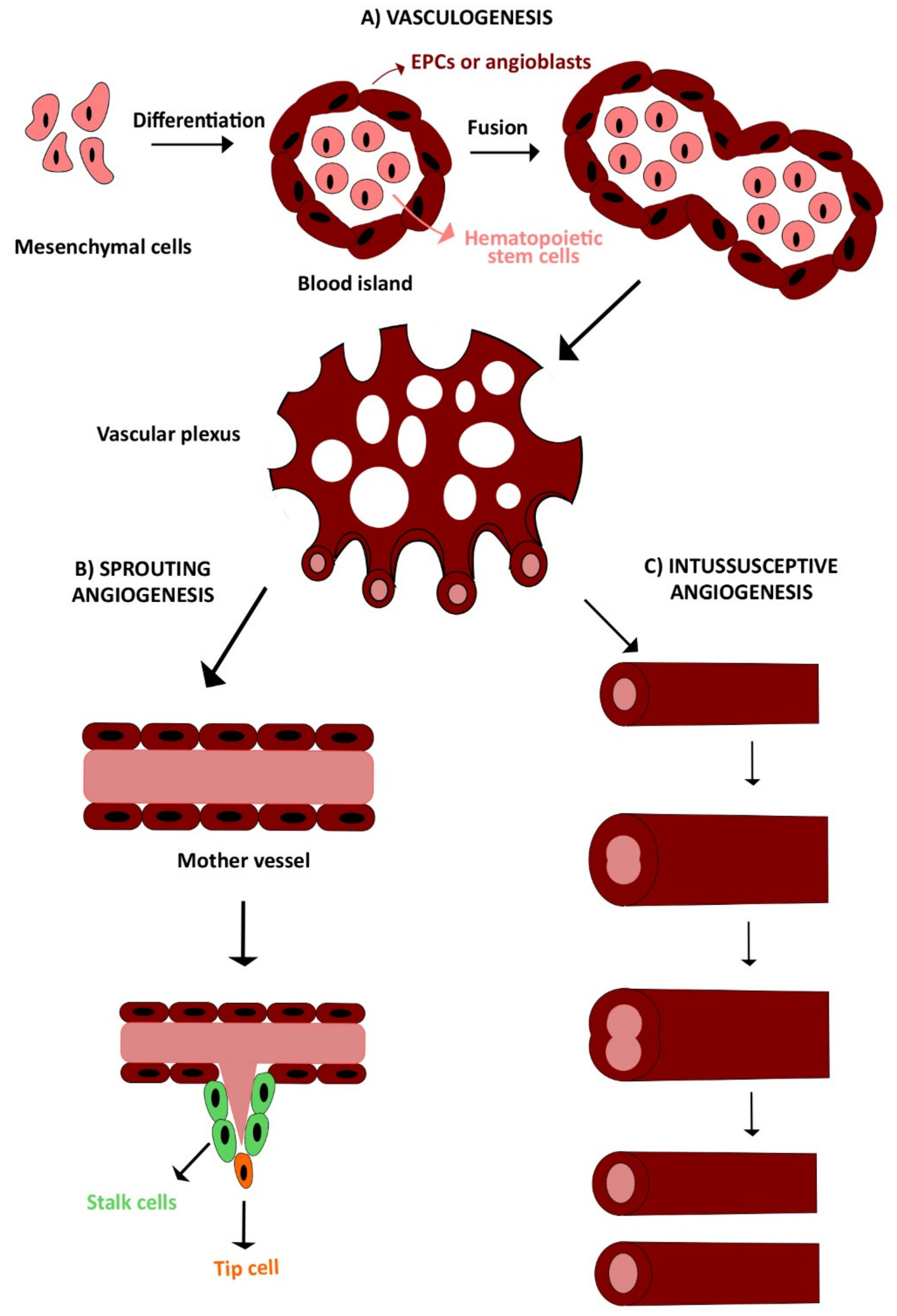
The development of the circulatory system requires the interplay of several finely regulated processes. There are three main mechanisms of vessel growth during embryonal and postnatal life: vasculogenesis, angiogenesis, and arteriogenesis (Figure 1) [3,9]. Vasculogenesis is defined as the de novo formation of vessels from aggregated endothelial precursors, while angiogenesis consists in neovessel formation from preexisting blood vessels and may be distinguished into sprouting angiogenesis and intussusceptive angiogenesis. The stabilization of these new sprouts by mural cells, e.g., pericytes and vascular smooth muscle cells (SMCs), is then termed arteriogenesis [9,49].
2.1. Vasculogenesis
Embryonal vasculogenesis represents one of the earliest events during organogenesis, as it starts already during gastrulation [50,51]. Vasculogenesis consists in the differentiation, expansion, and assembly of endothelial precursors into a functional blood vessel [49,52]. Vascular development in the embryo is initiated by mesodermal cells, also referred to as hemangioblasts, which migrate into the yolk sac, to form cellular aggregates known as blood islands. These are small clusters of round mesenchymal cells, which differentiate over time, thereby generating an outer layer of spindle-shaped endothelial precursors, i.e., angioblasts, and inner core of round primitive hematopoietic cells. Hence, angioblasts form sprouts and coalesce with adjacent blood islands to assembly into the early vascular plexus, which in turn generates primitive blood vessels (Figure 1A) [53,54].
2.2. Angiogenesis
Once the primitive vascular labyrinth has been built up, blood vessels are finely remodeled, to define tissues structures based on metabolic need, in a hierarchical tree-like network of arteries, arterioles, capillary beds, venules, and veins [19,49,55]. There are two angiogenic mechanisms: sprouting angiogenesis (Figure 1B) and intussusceptive angiogenesis (Figure 1C). Sprouting angiogenesis consists of neovessel formation from a preexisting capillary in response to a decrease in local oxygen tension [3,56]. It is characterized by different steps: vasodilation, increased vascular permeability, endothelial cell proliferation, and migration. In response to an angiogenic signal, e.g., VEGF, a leading tip cell is allowed to migrate in the cellular matrix after the degradation of basement membrane [57]. Thereafter, trailing endothelial stalk cells start to proliferate, determining sprout elongation and the trunk of new capillaries, without interrupting the connection with the parental vessel. Sprouts emerging from different vessels anastomose to constitute a functional vessel loop, allowing blood to flow, which also contributes to arterial–venous specification of endothelial cells [3,56]. Finally, the deposition of extracellular matrix and the recruitment of mural cells (e.g., pericytes and smooth muscle cells) leads to vessel maturation and stabilization. When the angiogenic signal is ceased, and upon restoration of the oxygen supply, endothelial cells return to a quiescent state, adopting their typical cobblestone-like shape [10,58]. The complexity and vascular surface of the capillary network can be further expanded by the insertion of a multitude of tissue pillars, according to a process observed for the first time in pulmonary capillary bed of neonatal rats [59] and referred to as intussusceptive (non-sprouting) angiogenesis [57].
2.3. Definition of Endothelial Progenitor Cells (EPCs) in the Adult: Myeloid Angiogenic Cells (MACs) and Endothelial Colony Forming Cells (ECFCs)
It has long been thought that sprouting angiogenesis represents the most important mechanism responsible for vessel growth and remodeling in postnatal life [3,9]. This dogma has been challenged in 1997 by Jeffrey Isner’s group, who reported, for the first time, the existence of circulating EPCs in peripheral blood of adult individuals [60]. A surge of investigations rapidly revealed that EPCs were mobilized from stem cell niches located in the endothelial intima of blood vessels and in bone marrow, to replace damaged/senescent endothelial cells and to rescue the vascular networks injured by an ischemic insult, thereby favoring tissue revascularization and rescuing local blood perfusion [6,18,61,62,63,64]. We refer the reader to several recent and exhaustive review articles which summarize the debate originated around the definition, classification, and characterization of the multiple EPC subtypes identified in peripheral circulation [6,65,66]. A recent consensus statement on nomenclature discouraged the indiscriminate use of the term EPC, as referred to the endothelial precursors circulating in the blood stream, in favor of a more stringent classification based on the cellular immunophenotype and pro-angiogenic activity [65]. Two intrinsically distinct lineages of cells encompass the multitude of EPC subtypes described in the literature: MACs, also known as “early” EPCs, and ECFCs, or late-outgrowth EPCs. MACs represent bone-marrow-derived hematopoietic cells which cooperate with ECFCs and support vascular repair and tissue regeneration through paracrine signaling [64]. ECFCs represent truly endothelial precursors, due to their high clonogenic potential and ability to physically engraft within emerging neovessels. In addition, ECFCs assemble into bidimensional tube networks in vitro, give rise to patent blood vessels, and anastomose with the host vasculature in vivo [67,68,69]. Furthermore, ECFCs sustain tissue revascularization by supporting the vasoreparative activity of other progenitor and stem cells, such as mesenchymal stem [70] and progenitor [4] cells, and by releasing a host of neurotrophic mediators, including growth factors [5], exosomes [71], and microvesicles [72]. A growing number of independent studies confirmed that ECFC transplantation was able to induce neovascularization and rescue blood perfusion in ischemic diseases, including PAD, AMI, ischemic brain disease, and retinopathy. It has therefore been proposed that ECFCs represent the most suitable cellular substrate to effect therapeutic angiogenesis in ischemic patients [12].
2.4. The Signaling Pathways of Angiogenesis and Vasculogenesis: The Role of Endothelial Ca2+ Signaling
As mentioned earlier, angiogenesis and vasculogenesis are finely regulated mechanisms [73]. Under physiological conditions, there is a balance between proangiogenic stimuli (e.g., VEGF, bFGF, PDGF, SDF-1-α, and angiopoietin-1 (ANG-1)/Tie2), and anti-angiogenic molecules (e.g., thrombospondin-1, endostatin, tumstatin, vasohibin, and C-X-C motif chemokine 10 (CXCL10) [3,19]. VEGF promotes angiogenesis synergistically with SDF-1α [74]. The tyrosine kinase receptor (TRK) VEGFR2, also denoted as kinase insert receptor (KDR), is the main isoform whereby VEGF signals to vascular endothelial cells and ECFCs [75,76]. Upon VEGF binding, VEGFR2 dimerizes and undergoes transphosphorylation, thereby recruiting downstream angiogenic pathways [40,75,77]. VEGF, for example, activates the following: (1) the phospholipase Cγ (PLCγ)-extracellular signal-regulated kinases 1/2 (ERK1/2) pathway, which is a crucial player during vascular development and in adult arteriogenesis (Figure 2A); (2) the phosphoinositide 3-kinases(PI3K)–protein kinase B (Akt)-mammalian target of rapamycin (mTOR) pathway, which underlies cell survival and the regulation of vasomotion and barrier function; (3) endothelial nitric oxide (NO) synthase (eNOS), thereby leading to NO release, which regulates endothelial cell proliferation, migration, and neovessel formation; and (4) Src and small GTPases pathways, which regulate cell shape, migration, and polarization [75] (Figure 2B). In particular, Src phosphorylates focal adhesion kinase (FAK), which in turn is a crucial player in the regulation of cell shape and adhesion [78]. Moreover, Src induces the disruption of adherens junctions and an increase in vascular permeability through vascular endothelial (VE)-cadherin phosphorylation [79]. Likewise, the Gi-protein coupled receptor, CXCR4 mediates SDF-1α-induced endothelial motility by enrolling a wide array of intracellular signaling pathways, including PLCβ and PLCγ2, RAS-mitogen-activated protein kinase (MAPK), and PI3K-Akt-mTOR [27,80].
Another fundamental signal whereby VEGF and SDF-1α stimulate neovessel formation is an increase in intracellular Ca2+ concentration ([Ca2+]i) [18,22,27,32,35,81]. As mentioned in the Introduction, endothelial Ca2+ signals regulate all the crucial steps involved in angiogenesis and vasculogenesis, including decrease in permeability, proliferation, and migration [18,75]. A number of Ca2+-sensitive decoders mediate the pro-angiogenic outcome of endothelial Ca2+ signals: These include NFAT, NF-κB, RAS-MAPK, PI3K/Akt, eNOS, myosin light chain kinase (MLCK), Ca2+/calmodulin-dependent protein kinase II (CaMKII), and calpain [18,75]. VEGF and SDF-1α elicit a dynamic interplay between InsP3-dependent Ca2+ release and SOCE, to trigger multiple intracellular Ca2+ signatures [22,27,32,81]. Nevertheless, some TRP isoforms may support VEGF-induced extracellular Ca2+ entry to regulate angiogenesis and arteriogenesis [18,35]. For instance, TRPC3 mediates VEGF-induced Ca2+ entry in human dermal microvascular endothelial cells [82], HUVECs [83], and umbilical cord blood-derived ECFCs [26], whereas TRPM2 supports VEGF-induced extracellular Ca2+ entry in human pulmonary artery endothelial cells [84]. Additionally, endothelial TRP channels may stimulate neovessel formation by sensing changes in the local microenvironment. For instance, TRPM2 is also sensitive to reactive oxygen species (ROS) [84], whereas TRPC5 is stimulated by hypoxia, to induce sprouting angiogenesis [85]. In this view, TRPV1 is emerging as a novel regulator of angiogenesis by means of its ability to detect both pro-angiogenic cues (e.g., erythropoietin) and environmental inputs (e.g., heat and ROS).
3. TRPV1: Molecular Structure and Gating Mechanisms
TRPV1 represents the funding member of the TRPV subfamily. TRP channels are featured by six transmembrane (TM1-6) α-helix segments, cytosolic NH2-, and COOH-tails and present a cation-permeable region formed by a loop between TM5 and TM6 [35]. The NH2- and COOH- terminals present a wide variability inside the superfamily being featured by different functions and lengths. In some members, they may interact with regulatory domains of cytosolic proteins (e.g., kinases, cytoskeletal proteins, and Ca2+-dependent sensors) [36,86]. However, there are two highly conserved regions inside the TRP domain (TRP-box1 and TRP-box2) that limit a central sequence characterized by a high degree of variability [87]. Surprisingly, the TRP-box2 is absent in the TRPV subfamily, although this shows a TRP-like domain, whose α-helix configuration and role resemble those of canonical TRP domains [35]. Additionally, TRPV channels share multiple ankyrin repeat domains (ARDs) localized on NH2-terminal, which are responsible of several functions, such as cytoskeleton interactions and channel opening by membrane deformation and channel tetramerization [86]. TRPV subunits can assemble into homo- and heterotetramers, although it is not clear which combinations are present in the vascular system [34]. It has long been known that TRPV1 and TRPV4 subunits form heteromeric channel exclusively located on the plasma membrane [88]. Accordingly, TRPV4 was found to associate with TRPV1 into a functional heteromeric channel complex in mouse primary retinal microvascular endothelial cells [89]. Likewise, TRPV5 and TRPV6 may assemble into heteromeric complexes in the kidney and gastrointestinal tract, but they are yet to be reported in vascular endothelium [37,90]. Furthermore, TRPV subunits may physically interact with TRP channels from different subfamilies: For instance, TRPP2/TRPV4 [91], TRPC1/TRPV6 [92], TRPML3/TRPV5 [93], and TRPV4/TRPC6 [94] heteromeric combinations were reported in naïve cells from multiple tissues. Of note, TRPV4/TRPC1 can assemble into functional channels into vascular endothelial cells from different vascular districts [18], including human umbilical vein endothelial cells (HUVECs) [95], rabbit mesenteric artery endothelial cells [96], and mouse aortic endothelial cells [97], whereas TRPV4 has been shown to interact with TRPC1 and TRPP2, to form a flow-sensitive mechanosensor in naïve vascular endothelial cells [98].
3.1. TRPV1 Structure
TRPV1 has been the first TRP channel to be solved at cryo-EM resolution [99,100]. This analysis confirmed that each TRPV1 subunit consists of six TM domains arranged around a central ion conduction pathway contributed by TM5 and TM6 and the re-entrant pore loop between them. The highly conserved TRP domain forms an interfacial helix that is positioned at the COOH-terminus of TM6 and interacts with the pre-TM1 helix and the cytosolic linker between TM4 and TM5, thereby enabling channel ligands to allosterically modulate pore conformation. The outer mouth of TRPV1 tetramers is remarkably wide, although it is followed by a dual-gate channel pore presenting two restriction points at G643 (4.6 Å, the selectivity filter) and at I679 (5.3 Å, the hydrophobic lower gate) that are closed in the non-conducting state. However, TRPV1 activation requires major structural rearrangements in the ion conduction pore, including the re-entrant loop, the selectivity filter, and the lower gate. Accordingly, capsaicin, which is a potent activator of the channel, binds to a cytosolic pocket in proximity of the inner mouth and gates TRPV1 by pulling TM6 from adjacent subunits apart and dilating the lower gate, while the constriction at the selective filter is not relieved. Conversely, the spider toxin binds to an extracellular site near to the re-entrant pore and gates TRPV1 by tilting the pore helix away from the central axis of the ion conduction pathway, thereby widening the selectivity filter from 4.6 Å to 7.6 Å. At the same time, the channel pore is further expanded by disruption of the hydrogen bonds bridging the loops that follow TM5 and precede TM6 [99,100].
3.2. TRPV1: Biophysical Properties and Gating Mechanisms
The TRPV1 channel, also named vanilloid receptor 1 (VR1), is the most studied and characterized TRPV member [101]. TRPV1 is a non-selective cation channel with an outwardly rectifying current–voltage (I–V) relationship with a negative slope conductance region at membrane potentials more negative than −70 mV [102]. TRPV1 is featured by similar permeabilities to Na+ and K+, but it is considerably more permeable to Ca2+ and Mg2+ (PCa/PNa = 9.6; PMg/PNa = 5), and displays a surprisingly high permeability to large cations [102,103]. Being mainly located on the plasma membrane, TRPV1 activation results in extracellular Ca2+ entry down the electrochemical gradient at physiological membrane potentials [102]. TRPV1 is a polymodal cation channel activated by several mechanical and chemical stimuli, such as extracellular protons, products of plants origin (e.g., capsaicin and gingerol), noxious heat (>42 °C), spider-derived vanillotoxins, and hydrogen sulphide (H2S) [18,101,104,105,106,107,108]. TRPV1 may also be gated by some endogenous agonists, including fatty acids conjugated with amines (e.g., N-arachidonylethanolamine (AEA, anandamide), N-arachidonoyldopamine (NADA), N-oleoylethanolamine (OLEA), N-arachidonolylserine, and various N-acyltaurines and N-acylsalsolinols), ATP, adenosine, polyamines (e.g., as spermines and spermidines), prostaglandins, and leukotriene B4 and pH < 5.9 [109,110,111,112,113,114]. Interestingly, an elegant study by Nilius’ group showed that TRPV1 is also sensitive to membrane depolarization and that heat and capsaicin can function as gating modifiers through voltage-to-current relationship shifts [115]. Indeed, at room temperature (22–23 °C), the voltage-dependent activation of TRPV1 requires a strong positive shift in the membrane potential (up to around +150 mV). Conversely, at higher temperatures, i.e., 40–45 °C, TRPV1 activation may already occur at −50 mV. This means that heat and capsaicin induce the channel to open at more physiological voltages [115]. It has been demonstrated that also extracellular protons act in a similar manner, thereby promoting TRPV1 activation at resting temperature, which provides is an additional explanation of TRPV1 involvement in inflammatory conditions [116]. Finally, TRPV1 undergoes desensitization, mainly through a Ca2+-dependent mechanism. Extracellular Ca2+ influx, mediated by TRPV1 itself, activates an inhibitory feedback signal by recruiting multiple signaling pathways. First, incoming Ca2+ could engage the Ca2+-dependent phosphatase, calcineurin, to counteract protein kinase C (PKC)- and protein kinase A (PKA)-dependent phosphorylation; second, Ca2+ could promote desensitization by binding to calmodulin [117].
4. The Physiological Role of TRPV1 in Vascular Endothelium
TRPV1 has been discovered in neuronal cells, being especially abundant in rat dorsal root ganglia (DRG) [101], and rapidly found also in trigeminal (TG) and nodose (NG) ganglia [107]. Several evidences until our days have shown a widely expression of TRPV1 also in non-neuronal cell, e.g., smooth muscular cells, keratinocytes, urothelium, liver, glial cells, mast cells, and macrophages [117,118,119]. Furthermore, TRPV1 is largely expressed in the intimal layer of blood vessels throughout peripheral vasculature, as reviewed in [119], and in umbilical cord blood (UCB)-derived [120] and circulating [40] ECFCs. The opening of TRPV1 in the plasma membrane causes the entry of extracellular Ca2+ down the electrochemical gradient, which therefore results in an increase in [Ca2+]i [121,122,123]. There is no evidence that TRPV1-mediated Ca2+ influx results in regenerative ER-dependent Ca2+ release, following the recruitment of ryanodine-receptors through the process of Ca2+-induced release, as observed in rat dorsal root ganglion neurons [124]. TRPV1 expression has also been found in the ER in several cell types, including rat nociceptive neurons, [125] and breast [126] and prostate [127,128] cancer cell lines. Cytosolic localization of TRPV1 has been recently described in bovine retinal microvascular endothelial cells (RMECs) [89]. It is, however, still unclear whether intracellular TRPV1 contributes to endothelial Ca2+ signaling.
4.1. TRPV1 Mediates Endothelium-Dependent Vasodilation
The interrelationship between TRPV1 and vascular endothelium was uncovered for the first time in 1999, when Zygmunt and co-workers proposed TRPV1 involvement in the regulation of vascular tone. They administered the powerful vasodilator, anandamide, to multiple isolated vascular preparations, including rat hepatic and small mesenteric arteries and guinea-pig basilar artery [129]. Anandamide was in turn found to activate TRPV1 channels localized on perivascular sensory fibers. TRPV1 activation induced the release of the vasodilator neuropeptide, calcitonin gene-related peptide (CGRP), which induced vasorelaxation by promoting endothelium-dependent hyperpolarization (EDH) [129]. Lo and co-workers provided one of the first pieces of evidence in favor of the role of endothelial TRPV1 in the control of vascular tone [130]. They indeed demonstrated that the vasorelaxant effect of VOA (N-(4-O-[2-methoxy, phenoxyethylaminobutyl]-3-methoxy benzyl)-nonamide), a synthetized molecule deriving from capsaicin, was endothelium-dependent [130]. First, they administered VOA to normotensive Wistar-Kyoto rats (WKY) and spontaneously hypertensive rats (SHR), thereby demonstrating a remarkable drop in systemic blood pressure. Then, they found that VOA was able to induce vasorelaxation in isolated rat thoracic aorta and mesenteric artery precontracted by phenylephrine (PE) in a TRPV1-dependent manner. Subsequently, they showed that VOA was able to induce CGRP release, followed by EDH in isolated vessel rings and to stimulate NO release in HUVECs [130]. It was, therefore, concluded that VOA was able to activate TRPV1, thereby promoting vasodilation in a CGRP/EDH and NO-dependent manner [130]. Two years later, Huidobro-Toro’s group reported that, although ineffective on vasorelaxation, 100 nM anandamide was able to promote robust NO release in the rat mesenteric bed by activating endothelial TRPV1 channels [131]. Anandamide-induced NO production was mimicked by TRPV1 agonists (i.e., resineferatoxin and capsaicin) and attenuated by TRPV1 antagonists (i.e., 5-iodoresiniferatoxin or SB 366791) [131]. An additional evidence for TRPV1-mediated endothelium-dependent vasorelaxation was provided by Zhu’s group in 2010 [132]. Capsaicin-induced TRPV1 activation induced the Ca2+-dependent phosphorylation of eNOS at Ser1177 in a PKA-dependent manner [132]. Consistently, long-term stimulation of TRPV1 induced vasorelaxation and lowered systemic blood pressure in SHR mice, accompanied by CGRP secretion from perivascular nerves. In this view, TRPV1 may be a therapeutic target in hypertension therapy in high-risk populations [132].
4.2. TRPV1 Ameliorates Endothelial Dysfunction in Diabetes, Atherosclerosis, and Metabolic Syndrome
Subsequent studies investigated whether and how TRPV1 could be targeted to mitigate endothelial dysfunction [133,134,135,136,137,138]. Earlier evidences showed that heat-stress-induced TRPV1 activation promoted CGRP secretion, thereby attenuating endothelial dysfunction induced by LPC, in mouse mesenteric artery and HUVECs [137,138]. Subsequently, Sun and collaborators analyzed TRPV1 involvement in oxidative-stress-induced endothelial dysfunction in diabetes, which represents one of the major cardiovascular risk factors. TRPV1 expression and PKA phosphorylation, as well as NO production, were decreased in porcine iliac artery endothelial cells maintained under high-glucose conditions [133]. Nonetheless, capsaicin rescued TRPV1 expression, thereby inducing the upregulation of Uncoupling Protein 2 (UCP2) in a PKA-dependent manner [133]. UCP2, in turn, ameliorated endothelial dysfunction by reducing the production of mitochondria-derived ROS and increasing NO release [133]. In agreement with these observations, capsaicin administration promoted UCP2 expression, mitigated ROS production, and restored NO-mediated, endothelium-dependent vasorelaxation in diabetic mice in vivo [133]. These data, therefore, demonstrated that TRPV1 may protect from hyperglycemia-induced endothelial dysfunction through the PKA/UCP2 pathway. Consistently, capsaicin stimulated endothelial TRPV1 to recruit the downstream PKA/UCP signaling cascade and thus mitigate mitochondrial dysfunction and restore coronary vasodilation and myocardial perfusion in atherosclerosis-prone apoliprotein E-deficient mice [134]. An additional elegant study proposed an alternative TRPV1-mediated mechanism of protection in endothelial dysfunction [135]. This investigation analyzed the effect of rutaecarpine, a major quinazolinocarboline alkaloid harvested from the Chinese herbal medicine, Evodia, on endothelial injury induced by Ox-LDL, a major risk factor for atherosclerosis [135]. Administration of Ox-LDL for 24 h decreased HUVEC viability and NO production, enhanced LDH release, and induced monocyte adhesion to the endothelial monolayer. However, endothelial damage was attenuated by pretreatment with rutaecarpine in a dose-dependent manner. Notably, capsazepine, a competitive TRPV1 antagonist, attenuated the beneficial effects of rutaecarpine on endothelial function [135]. Likewise, TRPV1 could effectively be targeted to treat metabolic syndrome [136,139]. Capsaicin-induced TRPV1 activation was found to mediate vasorelaxation by inducing NO release and EDH activation in coronary microcirculation from lean Ossabaw miniature swine [140]. Nevertheless, endothelial TRPV1 expression was significantly downregulated in obese animals, thereby compromising capsaicin-induced vasorelaxation [140]. Thus, it has been proposed that dietary agonists of endothelial TRPV1 could be employed to rescue coronary vasodilation in the presence of multiple cardiovascular risk factors, including diabetes, atherosclerosis, and metabolic syndrome [136,140]. Additionally, TRPV1 may protect vascular endothelial cells exposed to pro-inflammatory conditions associated, for instance, to lipopolysaccharide (LPS) [133,141]. Wang and collaborators demonstrated that TRPV1-activation suppressed LPS-induced HUVEC inflammation through eNOS activation and NF-kB inhibition [142]. Additionally, a TRPV1 protective effect was also reported in renal microvascular endothelial cells harvested from salt-sensitive hypertensive mice [142].
4.3. TRPV1 Regulates the Blood–Brain Barrier (BBB) Integrity
The only vascular district where endothelial TRPV1 has been associated to the impairment of vascular homeostasis is brain microcirculation. TRPV1 is largely expressed in human and mouse cerebromicrovascular endothelial cells [143,144]. A preliminary report showed that TRPV1 activation by anandamide reduced permeability in a human model of BBB by likely stimulating CGRP release [129,145]. A recent investigation disclosed that the endothelial expression of TRPV1 was enhanced within the peri-contusional region in a mouse model of traumatic brain injury (TBI) [143]. Pharmacological blockade of TRPV1 with capsazepine attenuated edema formation by preventing the dismantling of the BBB in vivo [143]. Subsequent evaluation of an in vitro model of BBB by using the mouse bEnd.3 cerebromicrovascular endothelial cell line confirmed that mechanical stimulation (imposed by biaxial stretch injury) caused endothelial cell apoptosis following TRPV1 activation [143]. Taken together, these findings demonstrate that endothelial TRPV1 plays a protective role under several pathological conditions, such as inflammation, diabetes, and atherosclerosis, while its role in brain ischemic remains to be determined. It, therefore, appears that endothelial TRPV1 could represent a useful target to prevent or attenuate endothelial dysfunction across peripheral vasculature.
5. Stimulating TRPV1 to Promote Angiogenesis
A growing number of evidences showed the involvement of TRPV1 in the angiogenic process [18,34,35,37]. However, capsaicin and piperine, both TRPV1 agonists, were initially found to inhibit angiogenesis both in vitro and in vivo [146,147]. It could be envisaged that chronic TRPV1 activation could lead to an aberrant increase in [Ca2+]i, thereby inducing to Ca2+-dependent cell death [148]. This hypothesis is supported by the notion that capsaicin-induced prolonged Ca2+ entry promotes apoptosis in prostate cancer cells [128], breast cancer cells [149], and cervical tumor cells [150]. On the other hand, TRPV1 activation was shown to reduce the ischemia/reperfusion (IR) injury in several organs, including the heart, brain, lungs, and kidney [151]. For instance, it has been demonstrated that TRPV1 activation during IR injury enhances substance P and CGRP from perivascular nerves and thereby afford cardioprotection [151,152,153]. It has also been suggested that TRPV1 activation may be induced upon myocardial injury by the reduction in local pH or by the proteases that are liberated from injured cells and activate the signaling cascade downstream of protease-activated receptor 2 (PAR2) [151,154,155]. Similarly, pharmacological activation of TRPV1 was found to induce neuroprotective effects, including a decrease in infarct volume and improvement of neurofunctional score, against IR injury in the brain [156,157]. It is, however, still unclear whether TRPV1 activation mediates revascularization upon ischemic insults in the heart and brain [151].
5.1. TRPV1 Sustains Angiogenesis Independently on VEGF
A recent study demonstrated that TRPV1 is able to trigger an angiogenic activity, even in the absence of parallel pro-angiogenic signaling pathways by VEGF. It was indeed shown that resiniferatoxin (RTX), a selective TRPV1 agonist, induced an outwardly rectifying non-selective cation current in primary cultures of bovine RMECs [89]. RTX-induced membrane currents were blocked by the selective TRPV1 inhibitors, capsazepine and A784168; furthermore, pharmacological blockade of TRPV1 with capsazepine and A784168 prevented the assembly of a bidimensional tube network in Matrigel scaffolds in the presence of 20% porcine serum [89]. Notably, these treatments did not affect either VEGF-induced intracellular Ca2+ signals or tubular network formation. These findings clearly demonstrated that TRPV1 activation per se is able to deliver a pro-angiogenic Ca2+ signal to vascular endothelial cells [89]. In agreement with this observation, blocking TRPV1 with capsazepine and A784168 inhibited neovascularization in a mouse model of oxygen-induced retinopathy [89]. Moreover, while pharmacological inhibition of TRPV1 with the selective antagonist SBB366791 did not prevent VEGF-induced tube formation in HUVEC, genetic deletion of TRPV1 abrogated stromal neovascularization in an in vivo cornea after a cauterization injury at the central cornea [158]. Consistently, the expression of pro-inflammatory/angiogenic factors (i.e., transforming growth factor β1 (TGFβ1) and VEGF) was suppressed in TRPV1-knockout (TRPV1−/−) mice [158]. Thus, targeting TRPV1 could provide an effective strategy to treat severe ischemic disorders. Of note, proximity ligation assay showed that TRPV1 could assemble with TRPV4 and blocking TRPV4 with selective blockers (i.e., HC06 and RN1734) also inhibited angiogenesis both in vitro and in vivo [89].
5.2. TRPV1 Stimulates Re-Endothelialization Following Vascular Injury
Apart from these sporadic observations, TRPV1 is widely recognized as a main trigger of angiogenesis. For instance, a recent investigation showed that TRPV1 was able to promote re-endothelialization after vascular injury [159]. These authors exploited the scratch wound assay to demonstrate that capsaicin increased HUVEC migration and proliferation in vitro by increasing mitochondrial energy metabolism and mitofusin (Mfn2) expression. In agreement with a role for TRPV1-mediated Ca2+ entry, the angiogenic effect of the dietary agonist capsaicin was inhibited by the TRPV1 antagonist, capsazepine, and by the membrane permeable Ca2+ chelator, BAPTA. These data were then therapeutically translated in vivo by performing wire injury of carotid artery in TRPV1-knockout (TRPV1−/−) mice and their wild-type (WT) littermates. Capsaicin was found to accelerate re-endothelialization and reduced neointimal formation in WT mice, but not in TRPV1 knockout mice. The regenerative effect of capsaicin was attenuated by genetic silencing of Mtfn2 both in vitro and in vivo [159], although the mechanism whereby TRPV1-mediated Ca2+ entry controls Mtfn2 expression is currently unclear. Of note, these data are consistent with the hypothesis that endothelial Ca2+ signaling could be properly manipulated to prevent restenosis, i.e., the reduction in lumen diameter, which occurs after percutaneous coronary intervention [160,161].
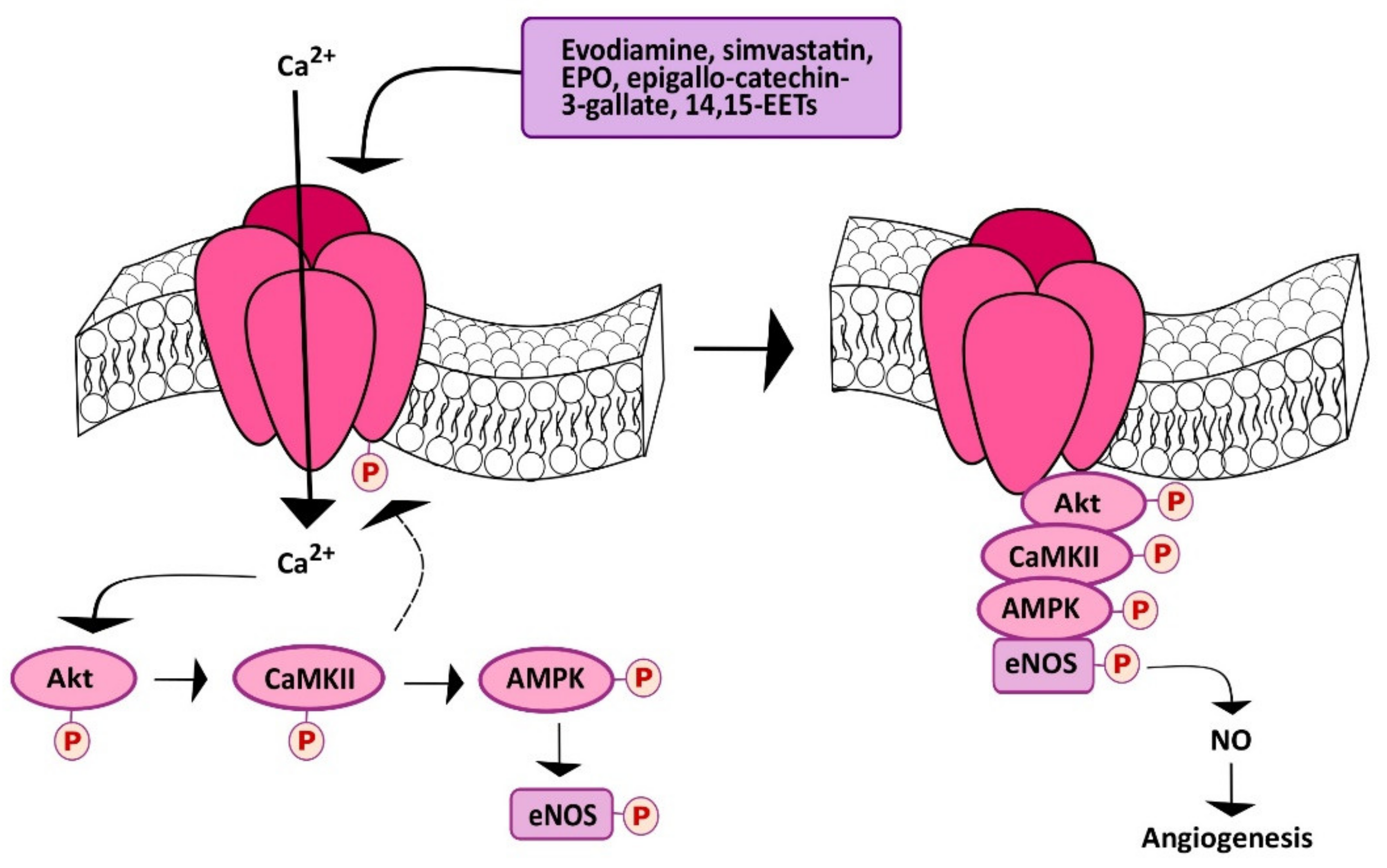
5.3. Chemical Stimulation of TRPV1 Promotes Angiogenesis in a Ca2+-Dependent Manner
Subsequent investigations demonstrated that TRPV1 could also be targeted to stimulate angiogenesis in a Ca2+-dependent manner. As described earlier, the first approach consisted of evaluating the effect of dietary or herbal TRPV1 agonists, such as capsaicin and evodiamine, respectively. Evodiamine is the major bioactive compound extracted from one of the most popular Chinese herbs, known as Wu-Chu-Yu (Evodiae fructus; Evodia rutaecarpa Benth., Rutaceae) [162]. Ching et al. investigated TRPV1-mediated eNOS activation and NO-dependent angiogenesis both in vitro and in vivo [163]. They found that evodiamine and capsaicin induced eNOS activation by phosphorylation and consequent NO release: Both of these effects were inhibited by pharmacological (with capsazepine) and genetic (with a specific small interfering RNA, siRNA) silencing of TRPV1. Evodiamine-induced TRPV1 activation was then found to recruit the Ca2+-dependent PI3K/Akt/CaMKII signaling pathway, which turned out to be necessary for ligand-induced phosphorylation of both TRPV1 and eNOS (Figure 3) [163]. Indeed, TRPV1 served as a scaffold for the recruitment and formation of a supermolecular complex consisting also of Akt, CaMKII and eNOS, which favored eNOS phosphorylation and NO release (Figure 4). This signaling pathway was also detected in mouse aortic endothelial cells (MAECs), in which genetic deletion of TRPV1 still prevented evodiamine from recruiting the PI3K/Akt/CaMKII/eNOS signaling cascade [163]. Of note, intraperitoneally injected evodiamine increased eNOS, Akt, and CaMKII phosphorylation in WT, but not TRPV1−/− mice. NO has long been known to promote neovascularization by stimulating both angiogenesis and vasculogenesis [136,164,165,166]. Consistently, the Matrigel plug assay confirmed that evodiamine promoted angiogenesis in vivo, although neovascularization was prevented in TRPV1−/− and eNOS-deficient (eNOS−/−) mice [163]. Of note, atherosclerotic lesions were more pronounced in ApoE-knockout mice (ApoE−/−), a widely employed animal model for hyperlipidemia, upon further deletion of TRPV1 (ApoE−/− TRPV1−/−). Likewise, evodiamine-induced phosphorylation of Akt, CaMKII, and eNOS was lower in ApoE−/−TRPV1−/−, as compared to TRPV1−/− mice [163]. A subsequent report further showed that evodiamine and capsaicin recruited AMP-activated protein kinase (AMPK) to phosphorylate eNOS in a CaMKII-dependent manner (Figure 4) [167]. Indeed, evodiamine also induced AMPK phosphorylation, but this effect was inhibited by blocking TRPV1 with capsazepine and CaMKII with the selective inhibitor KN62 [167]. Finally, evodiamine-induced eNOS phosphorylation was strongly reduced by compound C, a specific AMPK blocker, by overexpressing a dominant negative AMPK (dnAMPK) in Primary Bovine Aortic Endothelial Cells (BAECs). In agreement with these observations, AMPK activity proved to be essential for the ligand-induced physical association between TRPV1 and eNOS. As expected, pharmacological (with capsazepine) and/or genetic (with dnAMPK) blockade of AMPK also inhibited evodiamine-induced tube formation in Matrigel scaffolds both in vitro and in vivo [167]. Of note, this investigation demonstrated, for the first time, that TRPV1 could be effectively targeted to stimulate therapeutic angiogenesis. Intraperitoneal injection of evodiamine promoted neovascularization in a mouse model of hindlimb ischemia in an AMPK-dependent manner. Moreover, evodiamine reduced atherosclerotic plaques and increased phosphorylation of AMPK and eNOS in ApoE−/−, but not ApoE−/−TRPV1−/− mice [167]. These studies, therefore, strongly suggest that pharmacological stimulation of TRPV1 could represent an alternative strategy to induce therapeutic angiogenesis in ischemic tissues, even in the presence of established cardiovascular risk factors, e.g., hyperlipidemia.
The lipid-lowering drug simvastatin, which a 3-hydroxy-3-methylglutaryl-CoA reductase antagonist, is widely employed to reduce cholesterol biosynthesis and to reduce coronary artery disease events in subjects with or without diagnosed cardiovascular disease [168]. The beneficial effect of simvastatin on the cardiovascular system is strengthened by its documented ability to stimulate angiogenesis [169], trigger endothelium-dependent NO release [170,171], and improve left ventricular ejection fraction (LVEF) in heart failure [172]. Simvastatin has been shown to stimulate angiogenesis by causing VEGF expression following hypoxia-inducible factor-1α (HIF-1α) upregulation in vascular endothelial cells [169]. Furthermore, a recent report showed that simvastatin could also serve as TRPV1 agonist, thereby inducing NO release and NO-dependent angiogenesis [173]. Simvastatin (as well as lovastatin) was found to induce a rapid and prolonged (up to 240 min) increase in [Ca2+]i in Human Mammary Epithelial Cells (HMECs). The Ca2+ response to simvastatin was inhibited by two structurally unrelated TRPV1 inhibitors, such as capsazepine and SB366791; furthermore, simvastatin-induced intracellular Ca2+ signals were potentiated in HEK293 cells overexpressing a full-length TRPV1 WT, while it was inhibited by transducing the cells with TRPV1-Y671K (a TRPV1 mutant which is not able to conduct Ca2+) [173]. Similar to evodiamine, simvastatin stimulated TRPV1 to induce the Ca2+-dependent assembly of the TRPV1/Akt/CaMKII/AMPK/eNOS complex (Figure 4), thereby inducing eNOS phosphorylation, NO release, and NO-dependent tube formation in vitro [173]. Furthermore, simvastatin was able to promote neovascularization also in Matrigel plugs in vivo. Of note, simvastatin-induced NO production, tube formation in vitro, and neovascularization in vivo were inhibited by pharmacological (with capsazepine) or genetic (TRPV1−/− mice) blockade of TRPV1 [173]. In agreement with previous observations [169], NO may both induce HIF-1α expression [174] and stabilize HIF-1α activity [175]. It thus appears that TRPV1 activation represents an additional mechanism whereby statins (such as simvastatin and possibly lovastatin) exert a beneficial outcome on the cardiovascular system. Future pre-clinical studies are mandatory to assess whether simvastatin-induced TRPV1 activation can effectively be employed to stimulate therapeutic angiogenesis. Moreover, 14,15-Epoxyeicosatrienoic acid (14,15-EET) is a cytochrome P450 epoxygenase (CYP)-derived metabolite of arachidonic acid, which stimulates endothelial hyperpolarization [136], activates eNOS to induce NO release [176] (Figure 3), and stimulates endothelial cell migration and tube formation [177]. Pharmacological blockade of soluble epoxide hydroxylase (sHE), which is responsible for the rapid degradation of 14,15-EET into a less-active metabolite, has been put forward as an alternative mechanism to treat multiple cardiovascular disorders, such as atherosclerosis, hyperlipidemia, and hypertension [178]. A recent study demonstrated that 14,15-EET induced a rapid and prolonged (up to 60 min) increase in [Ca2+]i that disappeared in the absence of extracellular Ca2+ and upon pharmacological blockade of TRPV1 with capsazepine or SB366791 in HMEC (Figure 3) [121]. Similar to simvastatin, 14,15-EET-induced Ca2+ entry was, respectively, boosted or inhibited in HEK293 cells overexpressing a full-length TRPV1 WT or the TRPV1-Y671K with defective Ca2+ permeability. Thus, TRPV1 could be activated also by 14,15-EET, although through the intermediation of a GPCR [121], which is likely to be GRP40 [179]. Indeed, 14,15-EET-induced extracellular Ca2+ entry was blocked by the EET receptor blocker, 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EE5ZE), and a recent investigation disclosed that GRP40 activation resulted in a Ca2+ response similar to that evoked by 14,15-EET [179]. As reported above for evodiamine and simvastatin, 14.15-EET-induced extracellular Ca2+ entry then recruited eNOS to stimulate NO release and favor bidimensional tube formation in vitro and neovessel growth in Matrigel plugs in vivo (Figure 4) [121]. These findings, therefore, suggest that targeting sHE to promote 14,15-EET accumulation, thereby stimulating TRPV1-dependent pro-angiogenic Ca2+ signals, represents another promising strategy to treat ischemic disorders.
An additional approach to stimulate TRPV1-dependent angiogenesis is through dietary supplementation of epigallocatechin-3-gallate (EGCG) [122]. EGCG is one of the major effective components of green tea, by accounting for 50%–80% of its catechins [180]. Recent evidence suggested that green tea consumption could promote angiogenesis in the ischemic brain [181], whereas epidemiological studies and meta-analyses revealed that it could also confer protection against cardiovascular disease by stimulating endothelium-dependent NO release [182]. A recent investigation showed that EGCG was able to stimulate TRPV1 to induce NO release in BAECs [122]. EGCG induced a rapid and sustained (up to 240 min) elevation in [Ca2+]i that disappeared in the absence of extracellular Ca2+ and upon pharmacological blockade of TRPV1 with capsazepine or SB366791 (Figure 3). Moreover, EGCG-induced extracellular was, respectively, boosted or inhibited in HEK293 cells overexpressing a full-length TRPV1 WT or the TRPV1-Y671K with defective Ca2+ permeability [122]. Subsequent analysis confirmed that EGCG also stimulated TRPV1 to recruit the Ca2+-dependent Akt-CaMKII-AMPK signaling cascade, thereby leading to eNOS phosphorylation and NO release (Figure 4). Consistently, EGCG induced BAEC proliferation, migration, and tube formation through TRPV1 activation and NO release. Furthermore, EGCG promoted neovascularization in Matrigel plugs implanted in WT, but not TRPV1−/− mice [122]. These findings support the notion that tea green supplementation could be beneficial for cardiovascular patients, especially those affected by ischemic disorders. Finally, TRPV1 has been shown to support the pro-angiogenic effect of the erythroid growth factor, erythropoietin [123,183]. Erythropoietin stimulates an angiogenic behavior, e.g., proliferation, migration, and tube formation, both in vascular endothelial cells [184,185] and in circulating ECFCs; furthermore, the vasoreparative potential of ECFCs is enhanced by pretreatment with erythropoietin in an array of ischemic disorders [12], including hindlimb ischemia [186], cerebral ischemia [187], and retinopathy [89]. Notably, a recent investigation revealed that erythropoietin-induced angiogenesis requires TRPV1-mediated Ca2+ entry in BAECs [123]. By using the same approach previously described for simvastatin, 14,15-EET, and EGCG, e.g., pharmacological blockade with capsazepine and SBB366791 and genetically-mediated potentiation or inhibition with the full-length TRPV1 WT or the TRPV1-Y671K mutant defecting in Ca2+ permeation, EPO was found a sustained (up to 240 min) increase in [Ca2+]i [123]. Erythropoietin-induced intracellular Ca2+ signaling through TRPV1, in turn, recruited the Akt/AMPK signaling cascade to induce eNOS phosphorylation and NO release (Figure 4), thereby promoting tube formation in vitro and neovessel growth in Matrigel plugs implanted in WT, but not TRPV1−/− mice [123]. Of note, erythropoietin did not activate TRPV1 directly, but through the engagement of PLCγ1; indeed, pharmacological (with the selective blocker U73122 [188]) and genetic (via a selective siRNA) blockade of PLCγ1 prevented the Ca2+ response to erythropoietin by attenuating TRPV1 phosphorylation [123]. These findings, therefore, further support the notion that recombinant erythropoietin could be administrated to treat ischemic disorders and demonstrate that TRPV1 plays a pivotal role in the recruitment of the pro-angiogenic signaling pathways.
5.4. Is There a Role for TRPV1 in Heat-Induced Angiogenesis?
TRPV channels could confer temperature-sensing properties to vascular endothelial cells. For instance, TRPV4 may be activate by a temperature increase (from 19 to 38 °C) in mouse aortic endothelial cells [189]. A more recent paper suggested that TRPV1 could also sense heat increases to over 40 °C in human corneal endothelial cells [190]. The temperature sensibility of endothelial TRPV channels has been related to changes in NO release, which in turn regulates vascular tone and endothelial permeability [189,191]. An angiogenic stimulus could be triggered by whole-body hyperthermia (41.5–42.5 °C for 15 min, from a resting temperature of 37 °C) in rat cardiac myocardium. Likewise, far-infrared dry sauna (39 °C for 15 min followed by 34 °C for 20 min once daily for four weeks), also known as Waon therapy, promoted an increase in myocardial capillary density and attenuated cardiac hypertrophy in hypertensive rats [192]. The same approach was found to induce myocardial revascularization and attenuate maladaptive cardiac remodeling in a murine model of AMI [193]. Finally, near-infrared red exposure promoted capillary growth in the skin of human subjects, while locally increasing the temperature from 37 to 42 °C [194]. Of note, these procedures brought the whole-body and local temperature beyond the threshold for TRPV1 activation. The use of TRPV1−/− mice or the effect of Waon therapy in the presence of TRPV1 blockers are, however, required to assess TRPV1 involvement to the angiogenic switch observed in response to temperature increases.
6. TRPV1 Controls the Angiogenic Activity in ECFCs
A growing number of studies revealed the crucial role of Ca2+ signaling in circulating and umbilical-cord-blood-derived ECFCs [195,196]. Intracellular Ca2+ signals lie, indeed, at the center of an intricate network of signaling cascades that finely tune ECFC proliferation, migration, and neovessel formation [27,28,32,81]. For instance, VEGF elicits intracellular Ca2+ oscillations and promotes angiogenesis in circulating [32] and umbilical-cord-blood-derived ECFCs [26], whereas SDF-1α stimulates ECFC migration through a biphasic Ca2+ signal [27]. VEGF and SDF-1α trigger distinct intracellular Ca2+ signatures by inducing a concerted interplay between InsP3-induced ER Ca2+ release and SOCE [27,32]. However, TRPC3 has been shown to initiate the spiking Ca2+ response to VEGF in umbilical-cord-blood-derived ECFCs [26]. In addition, arachidonic acid, which is massively released in circulation by ischemic tissues [15], may also stimulate ECFCs to proliferate by activating TRPV4 followed by NO release [197]. The control of stem and progenitor cell fate is emerging as a compelling urgency for regenerative medicine [198]. In this regard, manipulating the signaling pathways that drive ECFC proliferation, migration, differentiation, and tubulogenesis could improve their regenerative outcome in vivo [6,12]. It has been proposed that Ca2+ signaling could be targeted to boost the regenerative potential of autologous ECFCs for regenerative purposes [15,47]. It has recently been demonstrated that TRPV1 is expressed and mediates extracellular Ca2+ entry in ECFCs [44,120]. Unlike bovine RMECs [89], TRPV1 is unlikely to interact with TRPV4 as the Ca2+ response to a specific TRPV4 agonist (GSK1016790A) was not sensitive to capsazepine [43]. Of note, TRPV1 controls ECFCs’ angiogenic activity both in a Ca2+-dependent and independent manner [44,120]; in particular, Lodola et al. provided the proof-of-concept that the optoceutical manipulation of TRPV1 is able to stimulate in vitro angiogenic [44], thereby paving the way for the therapeutic translation of this approach.
6.1. TRPV1 may Stimulate ECFC Proliferation in a Ca2+-Independent Manner
Anandamide is an endogenous endocannabinoid which may confer protection against multiple cardiovascular disorders, including hypertension and ischemia/reperfusion injury [199]. Earlier work showed that anandamide was not able per se to induce vasorelaxation in isolated rat mesenteric arteries, but it induced a rapid and transient NO production in endothelial cells that was inhibited by TRPV1 antagonists (e.g., 5-iodoresiniferatoxin, SB366791 or capsazepine) and mimicked by the TRPV1 agonists, RTX, and capsaicin [131]. As shown above, NO mediates TRPV1-dependent angiogenesis. Of note, a recent report demonstrated that anandamide entered into a HUVEC-derived cell line, i.e., EA.h926) and in ECFCs, through TRPV1, thereby stimulating proliferation and tube formation in a Ca2+-independent manner [120] (Figure 3). Indeed, anandamide uptake was inhibited by pharmacological (with SB366791) or genetic (via a siTRPV1) abrogation of TRPV1, while it was enhanced upon TRPV1 overexpression. Furthermore, capsaicin significantly attenuated anandamide uptake, which hints at a form of competition for the same binding site and is further consistent with TRPV1 engagement. Of note, anandamide uptake was not affected by removal of extracellular Ca2+, which seemingly ruled out the involvement of intracellular Ca2+ signaling in the downstream effects of TRPV1 [120]. Nevertheless, anandamide induced proliferation and tube formation in ECFCs, but this pro-angiogenic effect was abrogated by interfering with TRPV1 activity (with capsazepine) or expression (via a siTRPV1). It should be recalled that it has already been shown that TRP channels may support angiogenesis in a flux-independent manner [18]. For instance, genetic deletion of TRPC1 and TRPC4 inhibited proliferation, although it did not affect SOCE, in HUVECs [31]. Whatever the underlying mechanism, however, these preliminary data demonstrate that TRPV1 stimulates ECFC proliferation.
6.2. Gene-Less Opto-Stimulation of TRPV1 Leads to In Vitro Modulation of ECFC Fate
Pharmacological modulation of TRPV1 activity for regenerative purposes may suffer of important drawbacks that limit their applicative potential. More specifically, the injection of TRPV1 agonists, e.g., simvastatin, evodiamine, and erythropoietin, has limited spatial resolution and is often irreversible and does not allow temporally precise control.
Use of light as a stimulation tool recently emerged as a valid alternative, since it allows targeting single cells or even cellular sub-compartments, providing unprecedented spatial and temporal resolution, lower invasiveness, and higher selectivity. However, since the vast majority of mammalian cells do not bear any specific sensitivity to light, several strategies have been proposed to transduce light excitation into a biological stimulus. Among them, optogenetics, which regards the use of the genetic modification of cells to express light-sensitive actuators, is currently considered one of the most powerful methods for in vitro optical stimulation; still, the need for viral gene transfer raise important concerns toward its future clinical applicability [200]. For this reason, gene-less opto-stimulation of living cells and tissues is a growing field of research at the border among biophotonics, materials science, and biology [201].
Light-sensitive organic semiconductors are emerging as highly promising materials in biotechnology, thanks to a series of key enabling characteristics [202]: (i) they efficiently absorb light in the visible spectral range and undergo charge photogeneration; (ii) they sustain both electronic and ionic charge transport; (iii) they are soft, conformable, and solution processable; (iv) they can be easily tuned to enable specific excitation, probing, and sensing capabilities; and, most importantly, (v) they are highly biocompatible. Interestingly, a reliable optical modulation of the cell activity mediated by conjugated polymers has been reported in several previous reports, in vitro, at the level of single cells [203,204,205,206,207,208], ex vivo [209], and in vivo, as evidenced by behavioral studies on both invertebrate and vertebrate models [210,211].
Three different photostimulation mechanisms that are active at the polymer/cell interface have been proposed so far [202]. These include the following: (i) the creation of an interface capacitance, i.e., of a localized electric field, possibly affecting the cell membrane potential [212]; (ii) thermally mediated effects, which can modify the cell membrane electrical/mechanical/physical properties of the nearby cell [207,213]; (iii) photoactivated reduction/oxidation reactions occurring at the interface, leading to a local variation of extracellular and/or intracellular pH and sizable production of ROS at non-toxic concentration [202,207,214], and intracellular calcium modulation [215,216].
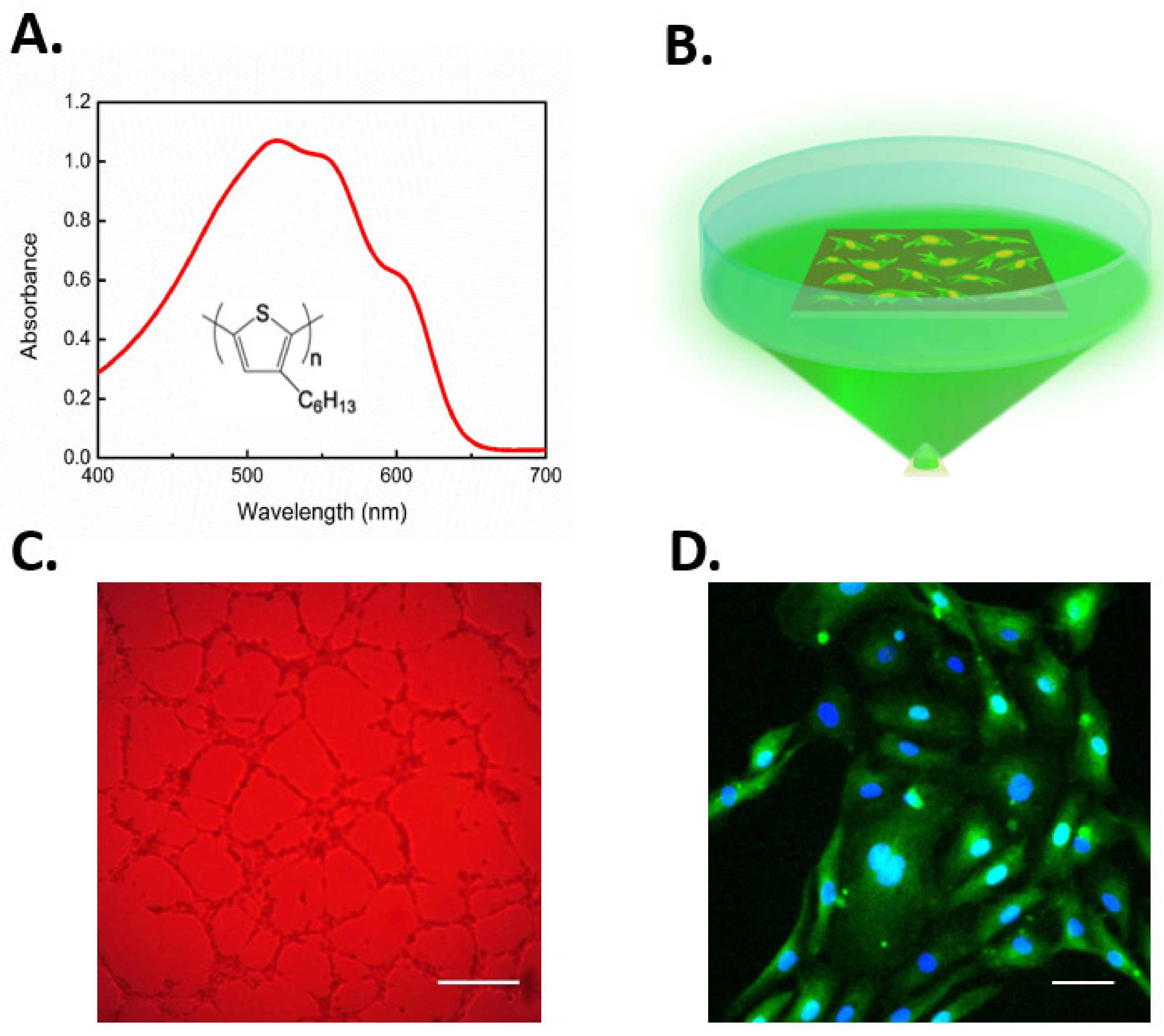
Recently, conjugated polymer semiconductors have also been used to gain optical control of cell fate via TRPV1 activation [207]. Lodola and colleagues demonstrated that rr-P3HT (regioregular poly(3-hexylthiophene))-mediated optical excitation during the first steps of ECFCs growth led to a robust increase of both proliferation and lumen formation in vitro (Figure 5A–C). Experimental evidences corroborated the biophysical scenario that optical excitation was able to stimulate ECFCs through TRPV1-mediated extracellular Ca2+ entry. This caused the downstream activation of Ca2+-dependent transcriptional factor NF-κB, thus leading to the expression of multiple pro-angiogenic genes that converted the initial optical excitation into an angiogenic response (Figure 5D). Of note, NF-κB is the Ca2+-dependent decoder that translates VEGF-induced intracellular Ca2+ oscillations into a pro-angiogenic output [32]. Finally, photostimulation-induced membrane depolarization was not sensitive to the pharmacological blockade of TRPV4, again arguing against the physical/functional interaction between TRPV1 and TRPV4. For the sake of completeness, the authors shone light also on the phototransduction mechanism demonstrating how the excitation of the TRPV1 channel through direct photothermal transduction seems not the predominant process, leading to enhanced tubulogenesis, thus pointing out how polymer surface photocatalytic activity played a major role in the phenomenon. Indeed, as mentioned earlier, ROS may activate TRPV1. It has, therefore, been suggested that optical stimulation of P3HT polymer induces the production of singlets and charge states, which in turn reduce oxygen dissolved in the medium to form superperoxide. Furthermore, superperoxide leads to the generation of hydrogen peroxide (H2O2), which can permeate the plasma membrane through specific aquaporins (e.g., aquaporin 3) and thereby activate TRPV1 [44,207,217]. Overall, this work evidenced how the combined use of optical excitation and organic polymers technology can open interesting perspectives for the modulation of progenitor cells fate and, more generally, corroborated the idea that controlling the signaling pathways driving ECFC proliferation, differentiation, and tube formation may represent a reliable strategy to improve the regenerative outcome of therapeutic angiogenesis.
7. Conclusions
Therapeutic angiogenesis represents one of the most promising strategies to rescue local blood flow in ischemic organs, when pharmacological treatment is ineffective and surgical vascularization is not feasible. Manipulating the intracellular signaling pathways, which drive endothelial proliferation, migration, and neovessel formation, could be crucial to enhance the clinical outcome of regenerative medicine. Therefore, unravelling novel pro-angiogenic mediators will enrich the arsenal of signaling pathways that can be targeted for therapeutic purposes. The polymodal TRPV1 channel is emerging as a crucial regulator of angiogenesis, due to its ability to integrate multiple chemical and physical stimuli, ranging from dietary agonists (e.g., capsaicin and EGCG) and the arachidonic acid-derivative 14,15-EET to a moderate increase in microenvironmental temperature or ROS levels. Furthermore, recent work demonstrated that some clinically available drugs, such as simvastatin or recombinant erythropoietin, could be repurposed to stimulate angiogenesis by activating TRPV1-mediated extracellular Ca2+ entry. The available evidence hints at vascular endothelial cells as the final actuators of agonists-induced TRPV1-dependent Ca2+ signals. Nevertheless, the evidence that TRPV1 is also expressed and mediates angiogenesis in circulating ECFCs strongly suggests that systemic or local administration of these small molecule drugs might also stimulate vasculogenesis. In this regard, a novel approach demonstrated that optical stimulation of TRPV1 could stimulate ECFCs with unprecedented spatial and temporal resolution and with no need of viral infection. Future work is necessary to assess whether this approach is also useful to stimulate capillary endothelial cells and to design a therapeutically relevant approach. This requires, e.g., the design of a strategy to precisely deliver visible light to the injured tissue or to exploit an infrared-sensitive photosensitive conjugated polymer, as well as the production of biocompatible and photosensitive nanoparticles to be injected within ischemic organs. Additional issues that remain to be clarified to better exploit TRPV1 for regenerative purposes are the following. First, it should be assessed whether TRPV1 supports VEGF-induced endothelial Ca2+ signals in vascular districts other than bovine retina. Likewise, TRPV1 contribution to the pro-angiogenic effect of other growth factors, such as bFGF and PDGF, should be evaluated. Second, it should be evaluated whether the intracellular distribution of TRPV1 is restricted to bovine RMECs or is a common feature in vascular endothelial cells and ECFCs. If so, the contribution of this intracellular TRPV1 pool to endothelial Ca2+ signaling and its Ca2+-dependent interaction with InsP3- and/or ryanodine-receptors should be investigated. Third, future work will have to show whether pharmacological or optoceutical stimulation of TRPV1 leads to revascularization also in the ischemic brain/heart. This issue is quite relevant due to the increasing incidence of brain stroke and AMI in the aging population. Fourth, TRPV1 may assemble with another pro-angiogenic Ca2+ entry pathway, such as TRPC1 [97,218], in response to ER Ca2+ depletion. It is, however, still unclear whether this signaling complex stimulates angiogenesis, an issue that surely deserves future investigation.
Author Contributions
Writing—original draft preparation, S.N.; writing—review and editing, V.R., F.L., and F.M.; editing, P.F. and M.R.A.; supervision, F.M. All the authors have read and agreed with the final version of this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022)—Department of Biology and Biotechnology “L. Spallanzani”, University of Pavia (F.M.), Fondo Ricerca Giovani from the University of Pavia (F.M.), and the EU Horizon 2020 FETOPEN-2018-2020 Program under Grant Agreement N. 828984, LION-HEARTED (M.R.A., F.L., and F.M.).
Acknowledgments
We do acknowledge Cecila Osera for the valuable assistance in LION-HEARTED management.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McCarron, J.G.; Lee, M.D.; Wilson, C. The Endothelium Solves problems that endothelial cells do not know exist. Trends Pharmacol. Sci. 2017, 38, 322–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarron, J.G.; Wilson, C.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Lee, M.D. Heterogeneity and emergent behaviour in the vascular endothelium. Curr. Opin. Pharmacol. 2019, 45, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banno, K.; Yoder, M.C. Tissue regeneration using endothelial colony-forming cells: Promising cells for vascular repair. Pediatr. Res. 2018, 83, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Paschalaki, K.E.; Randi, A.M. Recent Advances in endothelial colony forming cells toward their use in clinical translation. Front. Med. 2018, 5, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, C.L.; McLoughlin, K.J.; Chambers, S.E.J.; Guduric-Fuchs, J.; Stitt, A.W.; Medina, R.J. The Vasoreparative potential of endothelial colony forming cells: A journey through pre-clinical studies. Front. Med. 2018, 5, 273. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef]
- Poletto, V.; Rosti, V.; Biggiogera, M.; Guerra, G.; Moccia, F.; Porta, C. The role of endothelial colony forming cells in kidney cancer’s pathogenesis, and in resistance to anti-VEGFR agents and mTOR inhibitors: A speculative review. Crit. Rev. Oncol. Hematol. 2018, 132, 89–99. [Google Scholar] [CrossRef]
- Fischer, C.; Schneider, M.; Carmeliet, P. Principles and therapeutic implications of angiogenesis, vasculogenesis and arteriogenesis. Handb. Exp. Pharmacol. 2006, 157–212. [Google Scholar]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Madeddu, P. Therapeutic angiogenesis and vasculogenesis for tissue regeneration. Exp. Physiol. 2005, 90, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Tasev, D.; Koolwijk, P.; van Hinsbergh, V.W. Therapeutic potential of human-derived endothelial colony-forming cells in animal models. Tissue Eng. Part B Rev. 2016, 22, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Mitsos, S.; Katsanos, K.; Koletsis, E.; Kagadis, G.C.; Anastasiou, N.; Diamantopoulos, A.; Karnabatidis, D.; Dougenis, D. Therapeutic angiogenesis for myocardial ischemia revisited: Basic biological concepts and focus on latest clinical trials. Angiogenesis 2012, 15, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Qadura, M.; Terenzi, D.C.; Verma, S.; Al-Omran, M.; Hess, D.A. Concise review: Cell therapy for critical limb ischemia: An integrated review of preclinical and clinical studies. Stem Cells 2018, 36, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Berra-Romani, R.; Rosti, V. Manipulating intracellular Ca2+ signals to stimulate therapeutic angiogenesis in cardiovascular disorders. Curr. Pharm. Biotechnol. 2018, 19, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Tritto, S.; Signorelli, S.; Taglietti, V.; Tanzi, F. Epidermal growth factor induces intracellular Ca2+ oscillations in microvascular endothelial cells. J. Cell. Physiol. 2003, 194, 139–150. [Google Scholar] [CrossRef]
- Potenza, D.M.; Guerra, G.; Avanzato, D.; Poletto, V.; Pareek, S.; Guido, D.; Gallanti, A.; Rosti, V.; Munaron, L.; Tanzi, F.; et al. Hydrogen sulphide triggers VEGF-induced intracellular Ca(2+) signals in human endothelial cells but not in their immature progenitors. Cell Calcium 2014, 56, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial transient receptor potential channels and vascular remodeling: Extracellular Ca(2+) entry for angiogenesis, arteriogenesis and vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca(2+) signaling, angiogenesis and vasculogenesis: Just what it takes to make a blood vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef] [Green Version]
- Noren, D.P.; Chou, W.H.; Lee, S.H.; Qutub, A.A.; Warmflash, A.; Wagner, D.S.; Popel, A.S.; Levchenko, A. Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci. Signal. 2016, 9, ra20. [Google Scholar] [CrossRef] [Green Version]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, A.M.; Kurusamy, S.; Chen, Y.; Jiang, Z.; Chhabria, K.; MacDonald, R.B.; Kim, H.R.; Wilson, H.L.; van Eeden, F.J.M.; Armesilla, A.L.; et al. tmem33 is essential for VEGF-mediated endothelial calcium oscillations and angiogenesis. Nat. Commun. 2019, 10, 732. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Torres-Jácome, J.; Guzman-Silva, A.; Guerra, G.; Tanzi, F.; Moccia, F. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J. Vasc. Res. 2012, 49, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309. [Google Scholar] [CrossRef]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, A.D.; Riccardi, A.; et al. VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [CrossRef] [Green Version]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Guerra, G.; Borghesi, A.; Stronati, M.; Rosti, V.; Tanzi, F.; et al. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013, 22, 2561–2580. [Google Scholar] [CrossRef]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal cell-derived factor-1alpha promotes endothelial colony-forming cell migration through the Ca(2+)-dependent activation of the extracellular signal-regulated kinase 1/2 and phosphoinositide 3-kinase/AKT pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef]
- Balbi, C.; Lodder, K.; Costa, A.; Moimas, S.; Moccia, F.; van Herwaarden, T.; Rosti, V.; Campagnoli, F.; Palmeri, A.; De Biasio, P.; et al. Reactivating endogenous mechanisms of cardiac regeneration via paracrine boosting using the human amniotic fluid stem cell secretome. Int. J. Cardiol. 2019, 287, 87–95. [Google Scholar] [CrossRef]
- Dragoni, S.; Turin, I.; Laforenza, U.; Potenza, D.M.; Bottino, C.; Glasnov, T.N.; Prestia, M.; Ferulli, F.; Saitta, A.; Mosca, A.; et al. Store-operated Ca2+ entry does not control proliferation in primary cultures of human metastatic renal cellular carcinoma. Biomed. Res. Int. 2014, 2014, 739494. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic acid evokes an increase in Intracellular Ca(2+) concentration and nitric oxide production in endothelial cells from human brain microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smani, T.; Gomez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.M.; Rosado, J.A. TRP channels in angiogenesis and other endothelial functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakore, P.; Earley, S. Transient receptor potential channels and endothelial cell calcium signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [CrossRef]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The role of endothelial Ca(2+) signaling in neurovascular coupling: A view from the lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.C.; Gu, S.Y.; Bu, J.W.; Du, J.L. TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ. Res. 2010, 106, 1221–1232. [Google Scholar] [CrossRef] [Green Version]
- Lodola, F.; Laforenza, U.; Bonetti, E.; Lim, D.; Dragoni, S.; Bottino, C.; Ong, H.L.; Guerra, G.; Ganini, C.; Massa, M.; et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS ONE 2012, 7, e42541. [Google Scholar] [CrossRef] [Green Version]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient receptor potential channel expression signatures in tumor-derived endothelial cells: Functional roles in prostate cancer angiogenesis. Cancers 2019, 11, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanugula, A.K.; Adapala, R.K.; Midha, P.; Cappelli, H.C.; Meszaros, J.G.; Paruchuri, S.; Chilian, W.M.; Thodeti, C.K. Novel noncanonical regulation of soluble VEGF/VEGFR2 signaling by mechanosensitive ion channel TRPV4. FASEB J. 2019, 33, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragoni, S.; Guerra, G.; Fiorio Pla, A.; Bertoni, G.; Rappa, A.; Poletto, V.; Bottino, C.; Aronica, A.; Lodola, F.; Cinelli, M.P.; et al. A functional Transient Receptor Potential Vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J. Cell Physiol. 2015, 230, 95–104. [Google Scholar] [CrossRef]
- Lodola, F.; Rosti, V.; Tullii, G.; Desii, A.; Tapella, L.; Catarsi, P.; Lim, D.; Moccia, F.; Antognazza, M.R. Conjugated polymers optically regulate the fate of endothelial colony-forming cells. Sci. Adv. 2019, 5, eaav4620. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Martins, J.; Rondon-Clavo, C.; Tugal, D.; Korn, J.A.; Rizzi, R.; Padin-Iruegas, M.E.; Ottolenghi, S.; De Angelis, A.; Urbanek, K.; Ide-Iwata, N.; et al. Spontaneous calcium oscillations regulate human cardiac progenitor cell growth. Circ. Res. 2009, 105, 764–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, J.T.; Wagner, M.B.; Davis, M.E. Electrically induced calcium handling in cardiac progenitor cells. Stem Cells Int. 2016, 2016, 8917380. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca(2+) signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell Physiol. 2018, 233, 3901–3917. [Google Scholar] [CrossRef]
- Moccia, F.; Ruffinatti, F.A.; Zuccolo, E. Intracellular Ca(2+) signals to reconstruct a broken heart: Still a theoretical approach? Curr. Drug Targets 2015, 16, 793–815. [Google Scholar] [CrossRef]
- Udan, R.S.; Culver, J.C.; Dickinson, M.E. Understanding vascular development. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 327–346. [Google Scholar] [CrossRef]
- Goldie, L.C.; Nix, M.K.; Hirschi, K.K. Embryonic vasculogenesis and hematopoietic specification. Organogenesis 2008, 4, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Eichmann, A.; Yuan, L.; Moyon, D.; Lenoble, F.; Pardanaud, L.; Breant, C. Vascular development: From precursor cells to branched arterial and venous networks. Int. J. Dev. Biol. 2005, 49, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Iba, T.; Takakura, N. Mechanisms of new blood vessel formation and proliferative heterogeneity of endothelial cells. Int. Immunol. 2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risau, W.; Flamme, I. Vasculogenesis. Annu. Rev. Cell Dev. Biol. 1995, 11, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C. Endothelial stem and progenitor cells (stem cells): (2017 Grover Conference Series). Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Heinke, J.; Patterson, C.; Moser, M. Life is a pattern: Vascular assembly within the embryo. Front. Biosci. 2012, 4, 2269–2288. [Google Scholar] [CrossRef]
- Herbert, S.P.; Stainier, D.Y. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 551–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djonov, V.; Baum, O.; Burri, P.H. Vascular remodeling by intussusceptive angiogenesis. Cell Tissue Res. 2003, 314, 107–117. [Google Scholar] [CrossRef]
- Fitzgerald, G.; Soro-Arnaiz, I.; De Bock, K. The warburg effect in endothelial cells and its potential as an anti-angiogenic target in cancer. Front. Cell Dev. Biol. 2018, 6, 100. [Google Scholar] [CrossRef] [Green Version]
- Caduff, J.H.; Fischer, L.C.; Burri, P.H. Scanning electron microscope study of the developing microvasculature in the postnatal rat lung. Anat. Rec. 1986, 216, 154–164. [Google Scholar] [CrossRef]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef]
- Liao, S.; Luo, C.; Cao, B.; Hu, H.; Wang, S.; Yue, H.; Chen, L.; Zhou, Z. Endothelial progenitor cells for ischemic stroke: Update on basic research and application. Stem Cells Int. 2017, 2017, 2193432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Cinelli, M.; Bonetti, E.; Guerra, G.; Rosti, V. Endothelial progenitor cells support tumour growth and metastatisation: Implications for the resistance to anti-angiogenic therapy. Tumour. Biol. 2015, 36, 6603–6614. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.; Perrotta, F.; Testa, G. Circulating endothelial progenitor cells biology and regenerative medicine in pulmonary vascular diseases. Curr. Pharm. Biotechnol. 2018, 19, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Edwards, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; Alexander, M.Y. Endothelial progenitor cells: New targets for therapeutics for inflammatory conditions with high cardiovascular risk. Front. Med. 2018, 5, 200. [Google Scholar] [CrossRef]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef]
- Yoder, M.C. Human endothelial progenitor cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006692. [Google Scholar] [CrossRef]
- Yoder, M.C. Is endothelium the origin of endothelial progenitor cells? Arter. Thromb. Vasc. Biol. 2010, 30, 1094–1103. [Google Scholar] [CrossRef] [Green Version]
- Yoder, M.C.; Mead, L.E.; Prater, D.; Krier, T.R.; Mroueh, K.N.; Li, F.; Krasich, R.; Temm, C.J.; Prchal, J.T.; Ingram, D.A. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 2007, 109, 1801–1809. [Google Scholar] [CrossRef] [Green Version]
- Medina, R.J.; O’Neill, C.L.; Humphreys, M.W.; Gardiner, T.A.; Stitt, A.W. Outgrowth endothelial cells: Characterization and their potential for reversing ischemic retinopathy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5906–5913. [Google Scholar] [CrossRef] [Green Version]
- Rohban, R.; Pieber, T.R. Mesenchymal stem and progenitor cells in regeneration: Tissue specificity and regenerative potential. Stem Cells Int. 2017, 2017, 5173732. [Google Scholar] [CrossRef] [Green Version]
- Burger, D.; Vinas, J.L.; Akbari, S.; Dehak, H.; Knoll, W.; Gutsol, A.; Carter, A.; Touyz, R.M.; Allan, D.S.; Burns, K.D. Human endothelial colony-forming cells protect against acute kidney injury: Role of exosomes. Am. J. Pathol. 2015, 185, 2309–2323. [Google Scholar] [CrossRef] [PubMed]
- Cantaluppi, V.; Gatti, S.; Medica, D.; Figliolini, F.; Bruno, S.; Deregibus, M.C.; Sordi, A.; Biancone, L.; Tetta, C.; Camussi, G. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int. 2012, 82, 412–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D.; Crivellato, E. “Sprouting angiogenesis”, a reappraisal. Dev. Biol. 2012, 372, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.J.; Song, S.; Seo, H.R.; Shin, J.H.; Choi, S.C.; Park, J.H.; Yu, C.W.; Hong, S.J.; Lim, D.S. Human endothelial colony forming cells from adult peripheral blood have enhanced sprouting angiogenic potential through up-regulating VEGFR2 signaling. Int. J. Cardiol. 2015, 197, 33–43. [Google Scholar] [CrossRef]
- Abhinand, C.S.; Raju, R.; Soumya, S.J.; Arya, P.S.; Sudhakaran, P.R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal. 2016, 10, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, M.A.; Serrels, B.; Fincham, V.J.; Frame, M.C.; Carragher, N.O. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol. Cell Biol. 2004, 24, 8113–8133. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Padhan, N.; Sjostrom, E.O.; Roche, F.P.; Testini, C.; Honkura, N.; Sainz-Jaspeado, M.; Gordon, E.; Bentley, K.; Philippides, A.; et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat. Commun. 2016, 7, 11017. [Google Scholar] [CrossRef] [Green Version]
- Teixido, J.; Martinez-Moreno, M.; Diaz-Martinez, M.; Sevilla-Movilla, S. The good and bad faces of the CXCR4 chemokine receptor. Int. J. Biochem. Cell Biol. 2018, 95, 121–131. [Google Scholar] [CrossRef]
- Dragoni, S.; Reforgiato, M.; Zuccolo, E.; Poletto, V.; Lodola, F.; Ruffinatti, F.A.; Bonetti, E.; Guerra, G.; Barosi, G.; Rosti, V.; et al. Dysregulation of VEGF-induced proangiogenic Ca2+ oscillations in primary myelofibrosis-derived endothelial colony-forming cells. Exp. Hematol. 2015, 43, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.W.; James, A.F.; Foster, R.R.; Hancox, J.C.; Bates, D.O. VEGF activates receptor-operated cation channels in human microvascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1768–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrikopoulos, P.; Eccles, S.A.; Yaqoob, M.M. Coupling between the TRPC3 ion channel and the NCX1 transporter contributed to VEGF-induced ERK1/2 activation and angiogenesis in human primary endothelial cells. Cell Signal. 2017, 37, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Urao, N.; Hecquet, C.M.; Zhang, M.; Sudhahar, V.; Gao, X.P.; Komarova, Y.; Ushio-Fukai, M.; Malik, A.B. Novel role of reactive oxygen species-activated Trp melastatin channel-2 in mediating angiogenesis and postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 877–887. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Lu, Y.; Qu, C.; Miller, M.; Tian, J.; Thakur, D.P.; Zhu, J.; Deng, Z.; Hu, X.; Wu, M.; et al. Identification and optimization of 2-aminobenzimidazole derivatives as novel inhibitors of TRPC4 and TRPC5 channels. Br. J. Pharmacol. 2015, 172, 3495–3509. [Google Scholar] [CrossRef] [Green Version]
- Gaudet, R. TRP channels entering the structural era. J. Physiol. 2008, 586, 3565–3575. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Yang, F.; Takanishi, C.L.; Zheng, J. Thermosensitive TRPV channel subunits coassemble into heteromeric channels with intermediate conductance and gating properties. J. Gen. Physiol. 2007, 129, 191–207. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, C.; McGahon, M.K.; Ashraf, S.; McNaughten, J.; Friedel, T.; Cincola, P.; Barabas, P.; Fernandez, J.A.; Stitt, A.W.; McGeown, J.G.; et al. Involvement of TRPV1 and TRPV4 channels in retinal angiogenesis. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3297–3309. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Sun, C.; Zheng, J. Heteromerization of TRP channel subunits: Extending functional diversity. Protein Cell 2010, 1, 802–810. [Google Scholar] [CrossRef] [Green Version]
- Stewart, A.P.; Smith, G.D.; Sandford, R.N.; Edwardson, J.M. Atomic force microscopy reveals the alternating subunit arrangement of the TRPP2-TRPV4 heterotetramer. Biophys. J. 2010, 99, 790–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindl, R.; Fritsch, R.; Jardin, I.; Frischauf, I.; Kahr, H.; Muik, M.; Riedl, M.C.; Groschner, K.; Romanin, C. Canonical transient receptor potential (TRPC) 1 acts as a negative regulator for vanilloid TRPV6-mediated Ca2+ influx. J. Biol. Chem. 2012, 287, 35612–35620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Grimm, C.; Becker, L.; Ricci, A.J.; Heller, S. A novel ion channel formed by interaction of TRPML3 with TRPV5. PLoS ONE 2013, 8, e58174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldenberg, N.M.; Wang, L.; Ranke, H.; Liedtke, W.; Tabuchi, A.; Kuebler, W.M. TRPV4 is required for hypoxic pulmonary vasoconstriction. Anesthesiology 2015, 122, 1338–1348. [Google Scholar] [CrossRef]
- Ma, X.; Qiu, S.; Luo, J.; Ma, Y.; Ngai, C.Y.; Shen, B.; Wong, C.O.; Huang, Y.; Yao, X. Functional role of vanilloid transient receptor potential 4-canonical transient receptor potential 1 complex in flow-induced Ca2+ influx. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, H.Z.E.; Carlton-Carew, S.R.E.; Khan, D.M.; Zargaran, A.K.; Jahan, K.S.; Vanessa Ho, W.S.; Albert, A.P. Heteromeric TRPV4/TRPC1 channels mediate calcium-sensing receptor-induced nitric oxide production and vasorelaxation in rabbit mesenteric arteries. Vasc. Pharmacol. 2017, 96, 53–62. [Google Scholar] [CrossRef]
- Ma, X.; Cheng, K.T.; Wong, C.O.; O’Neil, R.G.; Birnbaumer, L.; Ambudkar, I.S.; Yao, X. Heteromeric TRPV4-C1 channels contribute to store-operated Ca(2+) entry in vascular endothelial cells. Cell Calcium 2011, 50, 502–509. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Ma, X.; Shen, B.; Huang, Y.; Birnbaumer, L.; Yao, X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014, 28, 4677–4685. [Google Scholar] [CrossRef] [Green Version]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, T.; Simon, S.A. TRPV1 Receptors and Signal Transduction. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; Liedtke, W.B., Heller, S., Eds.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Munns, C.H.; Chung, M.K.; Sanchez, Y.E.; Amzel, L.M.; Caterina, M.J. Role of the outer pore domain in transient receptor potential vanilloid 1 dynamic permeability to large cations. J. Biol. Chem. 2015, 290, 5707–5724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzhikandathil, E.V.; Wang, H.; Szabo, T.; Morozova, N.; Blumberg, P.M.; Oxford, G.S. Functional analysis of capsaicin receptor (vanilloid receptor subtype 1) multimerization and agonist responsiveness using a dominant negative mutation. J. Neurosci. 2001, 21, 8697–8706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geron, M.; Hazan, A.; Priel, A. Animal toxins providing insights into TRPV1 activation mechanism. Toxins 2017, 9, 326. [Google Scholar] [CrossRef] [Green Version]
- Patacchini, R.; Santicioli, P.; Giuliani, S.; Maggi, C.A. Pharmacological investigation of hydrogen sulfide (H2S) contractile activity in rat detrusor muscle. Eur. J. Pharmacol. 2005, 509, 171–177. [Google Scholar] [CrossRef]
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Siemens, J.; Zhou, S.; Piskorowski, R.; Nikai, T.; Lumpkin, E.A.; Basbaum, A.I.; King, D.; Julius, D. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 2006, 444, 208–212. [Google Scholar] [CrossRef]
- Vriens, J.; Appendino, G.; Nilius, B. Pharmacology of vanilloid transient receptor potential cation channels. Mol. Pharmacol. 2009, 75, 1262–1279. [Google Scholar] [CrossRef]
- Appendino, G.; Minassi, A.; Pagani, A.; Ech-Chahad, A. The role of natural products in the ligand deorphanization of TRP channels. Curr. Pharm. Des. 2008, 14, 2–17. [Google Scholar] [CrossRef]
- Huang, S.M.; Bisogno, T.; Trevisani, M.; Al-Hayani, A.; De Petrocellis, L.; Fezza, F.; Tognetto, M.; Petros, T.J.; Krey, J.F.; Chu, C.J.; et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 8400–8405. [Google Scholar] [CrossRef] [Green Version]
- McNamara, F.N.; Randall, A.; Gunthorpe, M.J. Effects of piperine, the pungent component of black pepper, at the human vanilloid receptor (TRPV1). Br. J. Pharmacol. 2005, 144, 781–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Blair, N.T.; Clapham, D.E. Camphor activates and strongly desensitizes the transient receptor potential vanilloid subtype 1 channel in a vanilloid-independent mechanism. J. Neurosci. 2005, 25, 8924–8937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahern, G.P.; Wang, X.; Miyares, R.L. Polyamines are potent ligands for the capsaicin receptor TRPV1. J. Biol. Chem. 2006, 281, 8991–8995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voets, T.; Droogmans, G.; Wissenbach, U.; Janssens, A.; Flockerzi, V.; Nilius, B. The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature 2004, 430, 748–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aneiros, E.; Cao, L.; Papakosta, M.; Stevens, E.B.; Phillips, S.; Grimm, C. The biophysical and molecular basis of TRPV1 proton gating. EMBO J. 2011, 30, 994–1002. [Google Scholar] [CrossRef]
- Vennekens, R.; Owsianik, G.; Nilius, B. Vanilloid transient receptor potential cation channels: An overview. Curr. Pharm. Des. 2008, 14, 18–31. [Google Scholar] [CrossRef] [Green Version]
- Caterina, M.J.; Julius, D. The vanilloid receptor: A molecular gateway to the pain pathway. Annu. Rev. Neurosci. 2001, 24, 487–517. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef]
- Hofmann, N.A.; Barth, S.; Waldeck-Weiermair, M.; Klec, C.; Strunk, D.; Malli, R.; Graier, W.F. TRPV1 mediates cellular uptake of anandamide and thus promotes endothelial cell proliferation and network-formation. Biol. Open 2014, 3, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Su, K.H.; Lee, K.I.; Shyue, S.K.; Chen, H.Y.; Wei, J.; Lee, T.S. Implication of transient receptor potential vanilloid type 1 in 14,15-epoxyeicosatrienoic acid-induced angiogenesis. Int. J. Biol. Sci. 2014, 10, 990–996. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.C.; Wei, J.; Su, K.H.; Chiang, A.N.; Zhao, J.F.; Chen, H.Y.; Shyue, S.K.; Lee, T.S. Transient receptor potential vanilloid type 1 is vital for (-)-epigallocatechin-3-gallate mediated activation of endothelial nitric oxide synthase. Mol. Nutr. Food Res. 2015, 59, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.B.; Su, K.H.; Kou, Y.R.; Guo, B.C.; Lee, K.I.; Wei, J.; Lee, T.S. Role of transient receptor potential vanilloid 1 in regulating erythropoietin-induced activation of endothelial nitric oxide synthase. Acta Physiol. 2017, 219, 465–477. [Google Scholar] [CrossRef]
- Huang, W.; Wang, H.; Galligan, J.J.; Wang, D.H. Transient receptor potential vanilloid subtype 1 channel mediated neuropeptide secretion and depressor effects: Role of endoplasmic reticulum associated Ca2+ release receptors in rat dorsal root ganglion neurons. J. Hypertens. 2008, 26, 1966–1975. [Google Scholar] [CrossRef] [Green Version]
- Karai, L.J.; Russell, J.T.; Iadarola, M.J.; Olah, Z. Vanilloid receptor 1 regulates multiple calcium compartments and contributes to Ca2+-induced Ca2+ release in sensory neurons. J. Biol. Chem. 2004, 279, 16377–16387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, C.; Cordova, C.; Marchant, I.; Zuniga, R.; Ochova, P.; Ramirez-Barrantes, R.; Gonzalez-Arriagada, W.A.; Rodriguez, B.; Olivero, P. Intracellular aggregated TRPV1 is associated with lower survival in breast cancer patients. Breast Cancer 2018, 10, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecze, L.; Blum, W.; Henzi, T.; Schwaller, B. Endogenous TRPV1 stimulation leads to the activation of the inositol phospholipid pathway necessary for sustained Ca(2+) oscillations. Biochim. Biophys. Acta 2016, 1863, 2905–2915. [Google Scholar] [CrossRef]
- Pecze, L.; Josvay, K.; Blum, W.; Petrovics, G.; Vizler, C.; Olah, Z.; Schwaller, B. Activation of endogenous TRPV1 fails to induce overstimulation-based cytotoxicity in breast and prostate cancer cells but not in pain-sensing neurons. Biochim. Biophys. Acta 2016, 1863, 2054–2064. [Google Scholar] [CrossRef] [Green Version]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.; Sorgard, M.; Di Marzo, V.; Julius, D.; Hogestatt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef]
- Lo, Y.C.; Hsiao, H.C.; Wu, D.C.; Lin, R.J.; Liang, J.C.; Yeh, J.L.; Chen, I.J. A novel capsaicin derivative VOA induced relaxation in rat mesenteric and aortic arteries: Involvement of CGRP, NO, cGMP, and endothelium-dependent activities. J. Cardiovasc. Pharmacol. 2003, 42, 511–520. [Google Scholar] [CrossRef]
- Poblete, I.M.; Orliac, M.L.; Briones, R.; Adler-Graschinsky, E.; Huidobro-Toro, J.P. Anandamide elicits an acute release of nitric oxide through endothelial TRPV1 receptor activation in the rat arterial mesenteric bed. J. Physiol. 2005, 568, 539–551. [Google Scholar] [CrossRef]
- Yang, D.; Luo, Z.; Ma, S.; Wong, W.T.; Ma, L.; Zhong, J.; He, H.; Zhao, Z.; Cao, T.; Yan, Z.; et al. Activation of TRPV1 by dietary capsaicin improves endothelium-dependent vasorelaxation and prevents hypertension. Cell Metab. 2010, 12, 130–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Pu, Y.; Wang, P.; Chen, S.; Zhao, Y.; Liu, C.; Shang, Q.; Zhu, Z.; Liu, D. TRPV1-mediated UCP2 upregulation ameliorates hyperglycemia-induced endothelial dysfunction. Cardiovasc. Diabetol. 2013, 12, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, S.; Wang, P.; Ma, L.; Gao, P.; Gong, L.; Li, L.; Li, Q.; Sun, F.; Zhou, X.; He, H.; et al. Ameliorating endothelial mitochondrial dysfunction restores coronary function via transient receptor potential vanilloid 1-mediated protein kinase A/Uncoupling protein 2 pathway. Hypertension 2016, 67, 451–460. [Google Scholar] [CrossRef]
- Peng, W.J.; Liu, Y.; Yu, Y.R.; Fu, Y.Q.; Zhao, Y.; Kuang, H.B.; Huang, Q.R.; He, M.; Luo, D. Rutaecarpine prevented dysfunction of endothelial gap junction induced by Ox-LDL via activation of TRPV1. Eur. J. Pharmacol. 2015, 756, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Berra-Romani, R. Targeting the endothelial Ca 2+ tool kit to rescue endothelial dysfunction in obesity associated-hypertension. Curr. Med. Chem. 2019, 27, 240–257. [Google Scholar] [CrossRef]
- Ye, F.; Deng, P.Y.; Li, D.; Luo, D.; Li, N.S.; Deng, S.; Deng, H.W.; Li, Y.J. Involvement of endothelial cell-derived CGRP in heat stress-induced protection of endothelial function. Vasc. Pharmacol. 2007, 46, 238–246. [Google Scholar] [CrossRef]
- Luo, D.; Zhang, Y.W.; Peng, W.J.; Peng, J.; Chen, Q.Q.; Li, D.; Deng, H.W.; Li, Y.J. Transient receptor potential vanilloid 1-mediated expression and secretion of endothelial cell-derived calcitonin gene-related peptide. Regul. Pept. 2008, 150, 66–72. [Google Scholar] [CrossRef]
- McCarty, M.F.; DiNicolantonio, J.J.; O’Keefe, J.H. Capsaicin may have important potential for promoting vascular and metabolic health. Open Heart 2015, 2, e000262. [Google Scholar] [CrossRef] [Green Version]
- Bratz, I.N.; Dick, G.M.; Tune, J.D.; Edwards, J.M.; Neeb, Z.P.; Dincer, U.D.; Sturek, M. Impaired capsaicin-induced relaxation of coronary arteries in a porcine model of the metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2489–H2496. [Google Scholar] [CrossRef] [Green Version]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, L.; Xu, H.; Liu, S.; Zhu, F.; Yan, F.; Shen, S.; Zhu, M. TRPV1 agonism inhibits endothelial cell inflammation via activation of eNOS/NO pathway. Atherosclerosis 2017, 260, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.X.; Jing, Y.; Liu, Y.L.; Xu, Z.M.; Yuan, F.; Wang, M.L.; Geng, Z.; Tian, H.L. Inhibition of transient receptor potential vanilloid 1 attenuates blood-brain barrier disruption after traumatic brain injury in mice. J. Neurotrauma 2019, 36, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Golech, S.A.; McCarron, R.M.; Chen, Y.; Bembry, J.; Lenz, F.; Mechoulam, R.; Shohami, E.; Spatz, M. Human brain endothelium: Coexpression and function of vanilloid and endocannabinoid receptors. Brain Res. Mol. Brain Res. 2004, 132, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Hind, W.H.; Tufarelli, C.; Neophytou, M.; Anderson, S.I.; England, T.J.; O’Sullivan, S.E. Endocannabinoids modulate human blood-brain barrier permeability in vitro. Br. J. Pharmacol. 2015, 172, 3015–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.K.; Han, K.Y.; Kim, E.C.; Kim, Y.M.; Lee, S.W.; Kim, O.H.; Kim, K.W.; Gho, Y.S.; Kwon, Y.G. Capsaicin inhibits in vitro and in vivo angiogenesis. Cancer Res. 2004, 64, 644–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doucette, C.D.; Hilchie, A.L.; Liwski, R.; Hoskin, D.W. Piperine, a dietary phytochemical, inhibits angiogenesis. J. Nutr. Biochem. 2013, 24, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F. Endothelial Ca(2+) signaling and the resistance to anticancer treatments: Partners in crime. Int. J. Mol. Sci. 2018, 19, E217. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.T.; Peters, A.A.; Tan, P.T.; Roberts-Thomson, S.J.; Monteith, G.R. Consequences of activating the calcium-permeable ion channel TRPV1 in breast cancer cells with regulated TRPV1 expression. Cell Calcium 2014, 56, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Barrantes, R.; Cordova, C.; Gatica, S.; Rodriguez, B.; Lozano, C.; Marchant, I.; Echeverria, C.; Simon, F.; Olivero, P. Transient receptor potential vanilloid 1 expression mediates capsaicin-induced cell death. Front. Physiol. 2018, 9, 682. [Google Scholar] [CrossRef] [Green Version]
- Randhawa, P.K.; Jaggi, A.S. A Review on Potential Involvement of TRPV1 Channels in Ischemia-Reperfusion Injury. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, D.H. TRPV1 gene knockout impairs postischemic recovery in isolated perfused heart in mice. Circulation 2005, 112, 3617–3623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, B.; Wang, D.H. N-oleoyldopamine, a novel endogenous capsaicin-like lipid, protects the heart against ischemia-reperfusion injury via activation of TRPV1. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H728–H735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, B.; Wang, D.H. Protease-activated receptor 2-mediated protection of myocardial ischemia-reperfusion injury: Role of transient receptor potential vanilloid receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1681–R1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadesi, S.; Nie, J.; Vergnolle, N.; Cottrell, G.S.; Grady, E.F.; Trevisani, M.; Manni, C.; Geppetti, P.; McRoberts, J.A.; Ennes, H.; et al. Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J. Neurosci. 2004, 24, 4300–4312. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; de Petrocellis, L.; Pryce, G.; Baker, D.; Guglielmotti, V.; Di Marzo, V. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 2006, 139, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Balasubramanian, A.; Marrelli, S.P. Pharmacologically induced hypothermia via TRPV1 channel agonism provides neuroprotection following ischemic stroke when initiated 90 min after reperfusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R149–R156. [Google Scholar] [CrossRef] [Green Version]
- Tomoyose, K.; Okada, Y.; Sumioka, T.; Miyajima, M.; Flanders, K.C.; Shirai, K.; Morii, T.; Reinach, P.S.; Yamanaka, O.; Saika, S. Suppression of in vivo neovascularization by the loss of TRPV1 in mouse cornea. J. Ophthalmol. 2015, 2015, 706404. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Zhang, Y.; He, K.; Wei, S.; Pei, H.; Wang, Q.; Yang, D.; Yang, Y. Activation of transient receptor potential vanilloid 1 accelerates re-endothelialization and inhibits neointimal formation after vascular injury. J. Vasc. Surg. 2017, 65, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Buccheri, D.; Piraino, D.; Andolina, G.; Cortese, B. Understanding and managing in-stent restenosis: A review of clinical data, from pathogenesis to treatment. J. Thorac. Dis. 2016, 8, E1150–E1162. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef]
- Yu, H.; Jin, H.; Gong, W.; Wang, Z.; Liang, H. Pharmacological actions of multi-target-directed evodiamine. Molecules 2013, 18, 1826–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ching, L.C.; Kou, Y.R.; Shyue, S.K.; Su, K.H.; Wei, J.; Cheng, L.C.; Yu, Y.B.; Pan, C.C.; Lee, T.S. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc. Res. 2011, 91, 492–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altaany, Z.; Moccia, F.; Munaron, L.; Mancardi, D.; Wang, R. Hydrogen sulfide and endothelial dysfunction: Relationship with nitric oxide. Curr. Med. Chem. 2014, 21, 3646–3661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-induced nitric oxide signaling: Mechanisms and therapeutic implications. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolo, E.; Bottino, C.; Diofano, F.; Poletto, V.; Codazzi, A.C.; Mannarino, S.; Campanelli, R.; Fois, G.; Marseglia, G.L.; Guerra, G.; et al. Constitutive store-operated Ca(2+) entry leads to enhanced nitric oxide production and proliferation in infantile hemangioma-derived endothelial colony-forming cells. Stem Cells Dev. 2016, 25, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Ching, L.C.; Chen, C.Y.; Su, K.H.; Hou, H.H.; Shyue, S.K.; Kou, Y.R.; Lee, T.S. Implication of AMP-activated protein kinase in transient receptor potential vanilloid type 1-mediated activation of endothelial nitric oxide synthase. Mol. Med. 2012, 18, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jeon, H.L.; Park, S.J.; Shin, J.Y. Effect of statins, metformin, angiotensin-converting enzyme inhibitors, and angiotensin II receptor blockers on age-related macular degeneration. Yonsei Med. J. 2019, 60, 679–686. [Google Scholar] [CrossRef]
- Nishimoto-Hazuku, A.; Hirase, T.; Ide, N.; Ikeda, Y.; Node, K. Simvastatin stimulates vascular endothelial growth factor production by hypoxia-inducible factor-1alpha upregulation in endothelial cells. J. Cardiovasc. Pharmacol. 2008, 51, 267–273. [Google Scholar] [CrossRef]
- Settergren, M.; Bohm, F.; Ryden, L.; Pernow, J. Cholesterol lowering is more important than pleiotropic effects of statins for endothelial function in patients with dysglycaemia and coronary artery disease. Eur. Heart J. 2008, 29, 1753–1760. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.P.; Zhao, J.F.; Lin, S.J.; Shyue, S.K.; Guo, B.C.; Lu, T.M.; Lee, T.S. Asymmetric dimethylarginine limits the efficacy of simvastatin activating endothelial nitric oxide synthase. J. Am. Heart Assoc. 2016, 5, e003327. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, M.J.; Cauthen, C.A.; Biondi-Zoccai, G.G.; Abbate, A.; Vrtovec, B.; Khan, B.V.; Vetrovec, G.W. Meta-analysis of randomized controlled trials of statins versus placebo in patients with heart failure. Am. J. Cardiol. 2009, 104, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Su, K.H.; Lin, S.J.; Wei, J.; Lee, K.I.; Zhao, J.F.; Shyue, S.K.; Lee, T.S. The essential role of transient receptor potential vanilloid 1 in simvastatin-induced activation of endothelial nitric oxide synthase and angiogenesis. Acta Physiol. 2014, 212, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Lee, H.J.; Kim, G.C.; Choi, J.H.; Hong, J.W. Plasma cupping induces VEGF expression in skin cells through nitric oxide-mediated activation of hypoxia inducible factor 1. Sci. Rep. 2019, 9, 3821. [Google Scholar] [CrossRef] [PubMed]
- Brune, B.; Zhou, J. The role of nitric oxide (NO) in stability regulation of hypoxia inducible factor-1alpha (HIF-1alpha). Curr. Med. Chem. 2003, 10, 845–855. [Google Scholar] [CrossRef]
- Campbell, W.B.; Fleming, I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflug. Arch. 2010, 459, 881–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheranov, S.Y.; Karpurapu, M.; Wang, D.; Zhang, B.; Venema, R.C.; Rao, G.N. An essential role for SRC-activated STAT-3 in 14,15-EET-induced VEGF expression and angiogenesis. Blood 2008, 111, 5581–5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romashko, M.; Schragenheim, J.; Abraham, N.G.; McClung, J.A. epoxyeicosatrienoic acid as therapy for diabetic and ischemic cardiomyopathy. Trends Pharmacol. Sci. 2016, 37, 945–962. [Google Scholar] [CrossRef]
- Park, S.K.; Herrnreiter, A.; Pfister, S.L.; Gauthier, K.M.; Falck, B.A.; Falck, J.R.; Campbell, W.B. GPR40 is a low-affinity epoxyeicosatrienoic acid receptor in vascular cells. J. Biol. Chem. 2018, 293, 10675–10691. [Google Scholar] [CrossRef] [Green Version]
- Clement, Y. Can green tea do that? A literature review of the clinical evidence. Prev. Med. 2009, 49, 83–87. [Google Scholar] [CrossRef]
- Bai, Q.; Lyu, Z.; Yang, X.; Pan, Z.; Lou, J.; Dong, T. Epigallocatechin-3-gallate promotes angiogenesis via up-regulation of Nfr2 signaling pathway in a mouse model of ischemic stroke. Behav. Brain Res. 2017, 321, 79–86. [Google Scholar] [CrossRef]
- Deka, A.; Vita, J.A. Tea and cardiovascular disease. Pharmacol. Res. 2011, 64, 136–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiese, K. Warming Up to New Possibilities with the Capsaicin Receptor TRPV1: mTOR, AMPK, and Erythropoietin. Curr. Neurovasc. Res. 2017, 14, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Kimakova, P.; Solar, P.; Solarova, Z.; Komel, R.; Debeljak, N. Erythropoietin and its angiogenic activity. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Maltaneri, R.E.; Schiappacasse, A.; Chamorro, M.E.; Nesse, A.B.; Vittori, D.C. Aquaporin-1 plays a key role in erythropoietin-induced endothelial cell migration. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118569. [Google Scholar] [CrossRef] [PubMed]
- Hache, G.; Garrigue, P.; Bennis, Y.; Stalin, J.; Moyon, A.; Cerami, A.; Brines, M.; Blot-Chabaud, M.; Sabatier, F.; Dignat-George, F.; et al. ARA290, a specific agonist of erythropoietin/CD131 heteroreceptor, improves circulating endothelial progenitors’ angiogenic potential and homing ability. Shock 2016, 46, 390–397. [Google Scholar] [CrossRef]
- Garrigue, P.; Hache, G.; Bennis, Y.; Brige, P.; Stalin, J.; Pellegrini, L.; Velly, L.; Orlandi, F.; Castaldi, E.; Dignat-George, F.; et al. Erythropoietin pretreatment of transplanted endothelial colony-forming cells enhances recovery in a cerebral ischemia model by increasing their homing ability: A SPECT/CT study. J. Nucl. Med. 2016, 57, 1798–1804. [Google Scholar] [CrossRef] [Green Version]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca(2+) signals and nitric oxide release in human brain microvascular endothelial cells. Cell Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Watanabe, H.; Vriens, J.; Suh, S.H.; Benham, C.D.; Droogmans, G.; Nilius, B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J. Biol. Chem. 2002, 277, 47044–47051. [Google Scholar] [CrossRef] [Green Version]
- Mergler, S.; Valtink, M.; Taetz, K.; Sahlmuller, M.; Fels, G.; Reinach, P.S.; Engelmann, K.; Pleyer, U. Characterization of transient receptor potential vanilloid channel 4 (TRPV4) in human corneal endothelial cells. Exp. Eye Res. 2011, 93, 710–719. [Google Scholar] [CrossRef]
- Mergler, S.; Valtink, M.; Coulson-Thomas, V.J.; Lindemann, D.; Reinach, P.S.; Engelmann, K.; Pleyer, U. TRPV channels mediate temperature-sensing in human corneal endothelial cells. Exp. Eye Res. 2010, 90, 758–770. [Google Scholar] [CrossRef]
- Ihori, H.; Nozawa, T.; Sobajima, M.; Shida, T.; Fukui, Y.; Fujii, N.; Inoue, H. Waon therapy attenuates cardiac hypertrophy and promotes myocardial capillary growth in hypertensive rats: A comparative study with fluvastatin. Heart Vessel. 2016, 31, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Sobajima, M.; Nozawa, T.; Shida, T.; Ohori, T.; Suzuki, T.; Matsuki, A.; Inoue, H. Repeated sauna therapy attenuates ventricular remodeling after myocardial infarction in rats by increasing coronary vascularity of noninfarcted myocardium. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H548–H554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Kim, Y.K.; Cho, K.H.; Chung, J.H. Infrared exposure induces an angiogenic switch in human skin that is partially mediated by heat. Br. J. Dermatol. 2006, 155, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Calcium signaling in endothelial colony forming cells in health and disease. Adv. Exp. Med. Biol 2020, 1131, 1013–1030. [Google Scholar] [CrossRef]
- Moccia, F.; Guerra, G. Ca(2+) signalling in endothelial progenitor cells: Friend or foe? J. Cell Physiol. 2016, 231, 314–327. [Google Scholar] [CrossRef]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vasc. Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef]
- Griffin, M.F.; Butler, P.E.; Seifalian, A.M.; Kalaskar, D.M. Control of stem cell fate by engineering their micro and nanoenvironment. World J. Stem Cells 2015, 7, 37–50. [Google Scholar] [CrossRef]
- Puhl, S.L. Cannabinoid-sensitive receptors in cardiac physiology and ischaemia. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118462. [Google Scholar] [CrossRef]
- Fenno, L.; Yizhar, O.; Deisseroth, K. The development and application of optogenetics. Annu. Rev. Neurosci. 2011, 34, 389–412. [Google Scholar] [CrossRef]
- Antognazza, M.R.; Martino, N.; Ghezzi, D.; Feyen, P.; Colombo, E.; Endeman, D.; Benfenati, F.; Lanzani, G. Shedding light on living cells. Adv. Mater. 2015, 27, 7662–7669. [Google Scholar] [CrossRef]
- Di Maria, F.; Lodola, F.; Zucchetti, E.; Benfenati, F.; Lanzani, G. The evolution of artificial light actuators in living systems: From planar to nanostructured interfaces. Chem. Soc. Rev. 2018, 47, 4757–4780. [Google Scholar] [CrossRef] [PubMed]
- Martino, N.; Bossio, C.; Vaquero Morata, S.; Lanzani, G.; Antognazza, M.R. Optical control of living cells electrical activity by conjugated polymers. J. Vis. Exp. 2016, e53494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feyen, P.; Colombo, E.; Endeman, D.; Nova, M.; Laudato, L.; Martino, N.; Antognazza, M.R.; Lanzani, G.; Benfenati, F.; Ghezzi, D. Light-evoked hyperpolarization and silencing of neurons by conjugated polymers. Sci. Rep. 2016, 6, 22718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaquero, S.; Bossio, C.; Bellani, S.; Martino, N.; Zucchetti, E.; Lanzani, G.; Antognazza, M.R. Conjugated polymers for the optical control of the electrical activity of living cells. J. Mater. Chem. B 2016, 4, 5272–5283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucchetti, E.; Zangoli, M.; Bargigia, I.; Bossio, C.; Di Maria, F.; Barbarella, G.; D’Andrea, C.; Lanzani, G.; Antognazza, M.R. Poly(3-hexylthiophene) nanoparticles for biophotonics: Study of the mutual interaction with living cells. J. Mater. Chem. B 2017, 5, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Martino, N.; Tullii, G.; Lanzani, G.; Antognazza, M.R. Conjugated polymers mediate effective activation of the Mammalian Ion Channel Transient Receptor Potential Vanilloid 1. Sci. Rep. 2017, 7, 8477. [Google Scholar] [CrossRef] [Green Version]
- Tullii, G.; Giona, F.; Lodola, F.; Bonfadini, S.; Bossio, C.; Varo, S.; Desii, A.; Criante, L.; Sala, C.; Pasini, M.; et al. High-aspect-ratio semiconducting polymer pillars for 3D cell cultures. ACS Appl. Mater. Interfaces 2019, 11, 28125–28137. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, D.; Antognazza, M.R.; Maccarone, R.; Bellani, S.; Lanzarini, E.; Martino, N.; Mete, M.; Pertile, G.; Bisti, S.; Lanzani, G.; et al. A polymer optoelectronic interface restores light sensitivity in blind rat retinas. Nat. Photonics 2013, 7, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Maya-Vetencourt, J.F.; Ghezzi, D.; Antognazza, M.R.; Colombo, E.; Mete, M.; Feyen, P.; Desii, A.; Buschiazzo, A.; Di Paolo, M.; Di Marco, S.; et al. A fully organic retinal prosthesis restores vision in a rat model of degenerative blindness. Nat. Mater. 2017, 16, 681–689. [Google Scholar] [CrossRef]
- Tortiglione, C.; Antognazza, M.R.; Tino, A.; Bossio, C.; Marchesano, V.; Bauduin, A.; Zangoli, M.; Morata, S.V.; Lanzani, G. Semiconducting polymers are light nanotransducers in eyeless animals. Sci. Adv. 2017, 3, e1601699. [Google Scholar] [CrossRef] [Green Version]
- Gautam, V.; Rand, D.; Hanein, Y.; Narayan, K.S. A polymer optoelectronic interface provides visual cues to a blind retina. Adv. Mater. 2014, 26, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Martino, N.; Feyen, P.; Porro, M.; Bossio, C.; Zucchetti, E.; Ghezzi, D.; Benfenati, F.; Lanzani, G.; Antognazza, M.R. Photothermal cellular stimulation in functional bio-polymer interfaces. Sci. Rep. 2015, 5, 8911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antognazza, M.R.; Abdel Aziz, I.; Lodola, F. Use of exogenous and endogenous photomediators as efficient ROS modulation tools: Results and perspectives for therapeutic purposes. Oxidative Med. Cell. Longev. 2019, 2019, 2867516. [Google Scholar] [CrossRef] [PubMed]
- Bossio, C.; Aziz, I.A.; Tullii, G.; Zucchetti, E.; Debellis, D.; Zangoli, M.; Di Maria, F.; Lanzani, G.; Antognazza, M.R. Photocatalytic activity of polymer nanoparticles modulates intracellular calcium dynamics and reactive oxygen species in HEK-293 cells. Front. Bioeng. Biotech. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Tullii, G.; Desii, A.; Bossio, C.; Bellani, S.; Colombo, M.; Martino, N.; Antognazza, M.R.; Lanzani, G. Bimodal functioning of a mesoporous, light sensitive polymer/electrolyte interface. Org. Electron. 2017, 46, 88–98. [Google Scholar] [CrossRef]
- Martinotti, S.; Laforenza, U.; Patrone, M.; Moccia, F.; Ranzato, E. Honey-mediated wound healing: H2O2 entry through AQP3 determines extracellular Ca(2+) influx. Int. J. Mol. Sci. 2019, 20, 764. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Lodola, F.; Dragoni, S.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Ca2+ signalling in endothelial progenitor cells: A novel means to improve cell-based therapy and impair tumour vascularisation. Curr. Vasc. Pharmacol. 2014, 12, 87–105. [Google Scholar] [CrossRef]
Figure 1.
The two main processes involved in vascular development: vasculogenesis and angiogenesis. (A) Schematic representation of vasculogenesis, which consists of de novo vessel formation from aggregated endothelial precursors (EPCs or angioblasts) assembled within the blood islands. Thereafter, multiple blood islands fuse together into the early vascular plexus, which in turn generates primitive blood vessels. (B,C) Angiogenesis consists of neovessel formation from preexisting blood vessels in response to pro-angiogenic signals. Angiogenesis may occur through two different mechanisms: sprouting angiogenesis and intussusceptive angiogenesis. See text for detailed explanation.
Figure 1.
The two main processes involved in vascular development: vasculogenesis and angiogenesis. (A) Schematic representation of vasculogenesis, which consists of de novo vessel formation from aggregated endothelial precursors (EPCs or angioblasts) assembled within the blood islands. Thereafter, multiple blood islands fuse together into the early vascular plexus, which in turn generates primitive blood vessels. (B,C) Angiogenesis consists of neovessel formation from preexisting blood vessels in response to pro-angiogenic signals. Angiogenesis may occur through two different mechanisms: sprouting angiogenesis and intussusceptive angiogenesis. See text for detailed explanation.

Figure 2.
Role of endothelial Ca2+ signaling angiogenesis and vasculogenesis. (A) VEGF binds to VEGFR2 and induces receptor dimerization and transphosphorylation. Activation of VEGFR2, in turn, leads to InsP3-dependent Ca2+ release, followed by SOCE activation (not shown). The ensuing increase in [Ca2+]i leads to the activation of Ca2+-dependent decoders, such as calmodulin, which in turn activates calcineurin and Ca2+-Calmodulin-dependent kinase II (CaMKII). Calcineurin dephosphorylates NFAT, thereby promoting its translocation into the nucleus, where it activates genes responsible for cell proliferation and migration. CaMKII, in turn, inhibits calcineurin activity, but stimulates angiogenesis by phosphorylating multiple targets (i.e., Akt and Src, not shown). In addition, the increase in [Ca2+]i promotes cytosolic protein kinase C (PKC) relocation toward the plasma membrane. Herein, PKC is activated by DAG to stimulate the extracellular signal-regulated kinases 1/2 (ERK1/2) phosphorylation cascade. (B) Schematic representation of the Src and PI3K-Akt signaling pathways activated by VEGF. Once activated, VEGFR2 activates Src and subsequent downstream pathways involved in cell shape, adhesion, permeability, and survival. Moreover, VEGFR2 indirectly activates PI3K, either by Src or by VE-cadherin. PI3K generates phosphatidylinositol-3,4,5-trisphosphate (PIP3), which activates Akt, followed by eNOS activation and NO release. Finally, Akt activates FOXO1, which translocates into the nucleus, where, together with NO, induces transcription of genes involved in cell survival, migration, proliferation, and tube formation.
Figure 2.
Role of endothelial Ca2+ signaling angiogenesis and vasculogenesis. (A) VEGF binds to VEGFR2 and induces receptor dimerization and transphosphorylation. Activation of VEGFR2, in turn, leads to InsP3-dependent Ca2+ release, followed by SOCE activation (not shown). The ensuing increase in [Ca2+]i leads to the activation of Ca2+-dependent decoders, such as calmodulin, which in turn activates calcineurin and Ca2+-Calmodulin-dependent kinase II (CaMKII). Calcineurin dephosphorylates NFAT, thereby promoting its translocation into the nucleus, where it activates genes responsible for cell proliferation and migration. CaMKII, in turn, inhibits calcineurin activity, but stimulates angiogenesis by phosphorylating multiple targets (i.e., Akt and Src, not shown). In addition, the increase in [Ca2+]i promotes cytosolic protein kinase C (PKC) relocation toward the plasma membrane. Herein, PKC is activated by DAG to stimulate the extracellular signal-regulated kinases 1/2 (ERK1/2) phosphorylation cascade. (B) Schematic representation of the Src and PI3K-Akt signaling pathways activated by VEGF. Once activated, VEGFR2 activates Src and subsequent downstream pathways involved in cell shape, adhesion, permeability, and survival. Moreover, VEGFR2 indirectly activates PI3K, either by Src or by VE-cadherin. PI3K generates phosphatidylinositol-3,4,5-trisphosphate (PIP3), which activates Akt, followed by eNOS activation and NO release. Finally, Akt activates FOXO1, which translocates into the nucleus, where, together with NO, induces transcription of genes involved in cell survival, migration, proliferation, and tube formation.

Figure 3.
TRPV1 channel in angiogenesis. TRPV1 stimulates angiogenesis in response to evodiamine, simvastatin, EPO, epigallo-catechin-3-gallate, and 14,15-EETS in a Ca2+-dependent manner. Conversely, extracellular anandamide may enter through TRPV1, thereby stimulating angiogenesis in a Ca2+-independent manner.
Figure 3.
TRPV1 channel in angiogenesis. TRPV1 stimulates angiogenesis in response to evodiamine, simvastatin, EPO, epigallo-catechin-3-gallate, and 14,15-EETS in a Ca2+-dependent manner. Conversely, extracellular anandamide may enter through TRPV1, thereby stimulating angiogenesis in a Ca2+-independent manner.

Figure 4.
Proposed molecular mechanism of eNOS stimulation after TRPV1 activation. Activation of TRPV1 increases Ca2+ influx, which in turn activates PI3K/Akt/CaMKII signaling, leading to increased TRPV1 and eNOS phosphorylation. In addition, TRPV1 may serve as a scaffold for the formation of a complex comprising Akt, AMPK, CaMKII, and eNOS. Protein interactions seem to be important in eNOS activation and NO release.
Figure 4.
Proposed molecular mechanism of eNOS stimulation after TRPV1 activation. Activation of TRPV1 increases Ca2+ influx, which in turn activates PI3K/Akt/CaMKII signaling, leading to increased TRPV1 and eNOS phosphorylation. In addition, TRPV1 may serve as a scaffold for the formation of a complex comprising Akt, AMPK, CaMKII, and eNOS. Protein interactions seem to be important in eNOS activation and NO release.

Figure 5.
(A) The rr-P3HT chemical structure and optical absorption of the thin film. (B) Experimental setup and optical excitation protocol performed for evaluation of polymer-mediated cell photoexcitation effects on cell fate. (C) Representative image of in vitro network formation in ECFCs seeded on P3HT and subjected to optical excitation; scale bar: 250 µM. (D) Representative image of immunofluorescence staining, showing enhanced light-induced NF-κB nuclear translocation. Cell nuclei are detected by DAPI (blue) while cytoplasmic p65 NF-κB subunit with a secondary chicken anti-rabbit Alexa(488)-conjugated antibody (green). Scale bar: 50 µM. Figures adapted from [44,207].
Figure 5.
(A) The rr-P3HT chemical structure and optical absorption of the thin film. (B) Experimental setup and optical excitation protocol performed for evaluation of polymer-mediated cell photoexcitation effects on cell fate. (C) Representative image of in vitro network formation in ECFCs seeded on P3HT and subjected to optical excitation; scale bar: 250 µM. (D) Representative image of immunofluorescence staining, showing enhanced light-induced NF-κB nuclear translocation. Cell nuclei are detected by DAPI (blue) while cytoplasmic p65 NF-κB subunit with a secondary chicken anti-rabbit Alexa(488)-conjugated antibody (green). Scale bar: 50 µM. Figures adapted from [44,207].

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells 2020, 9, 1341. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061341
AMA Style
Negri S, Faris P, Rosti V, Antognazza MR, Lodola F, Moccia F. Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells. 2020; 9(6):1341. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061341
Chicago/Turabian StyleNegri, Sharon, Pawan Faris, Vittorio Rosti, Maria Rosa Antognazza, Francesco Lodola, and Francesco Moccia. 2020. "Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis" Cells 9, no. 6: 1341. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061341
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.