
Effect of Chronic Pioglitazone Treatment on Hepatic Gene Expression Profile in Obese C57BL/6J Mice

Abstract
:1. Introduction
2. Results
2.1. Pioglitazone Ameliorated Systemic Insulin Resistance

2.2. Pioglitazone Exacerbated Hepatic Steatosis

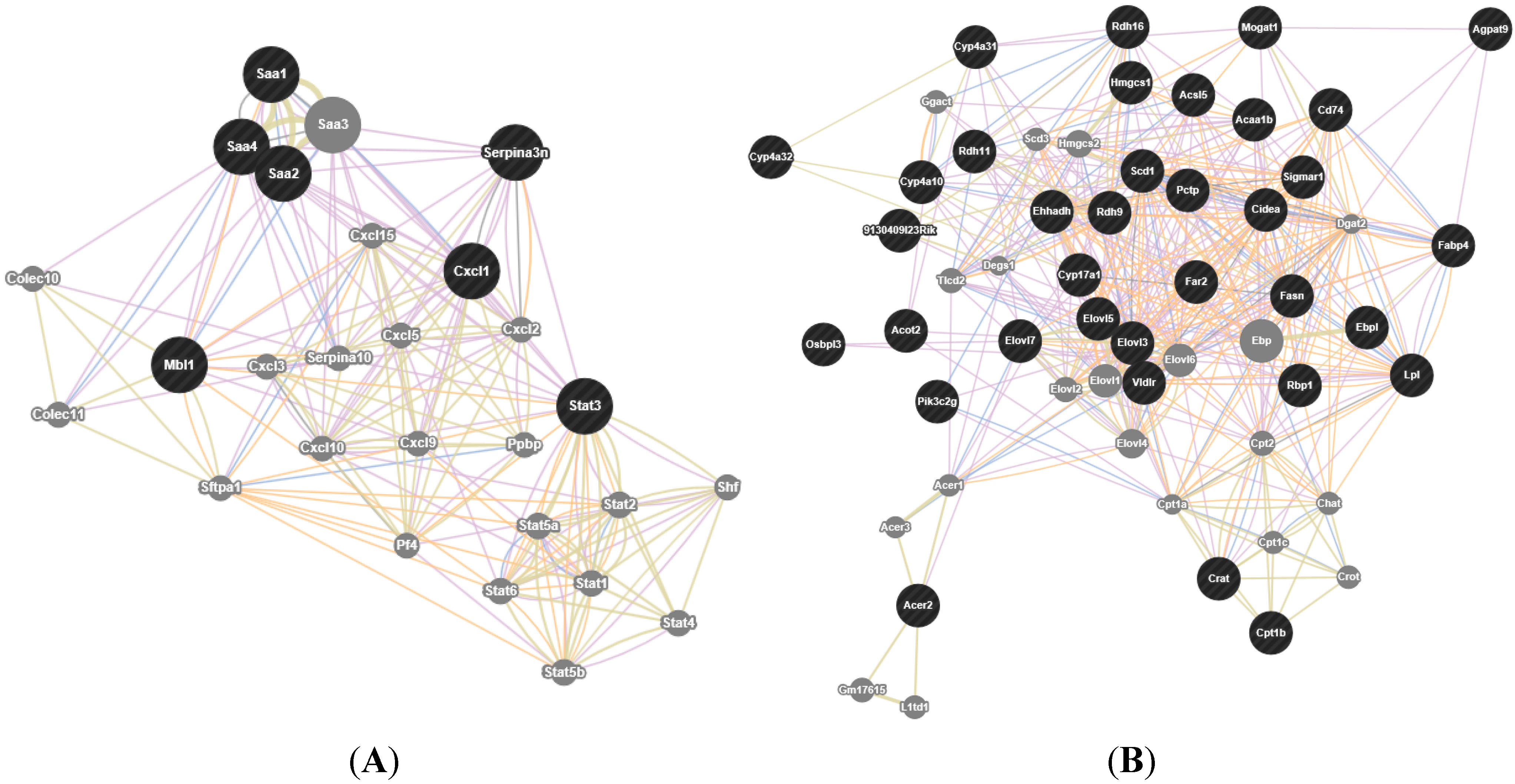
2.3. Identification and Classification of Differentially Expressed Genes by Microarray Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Function | Gene Count | Benjamini | Fold Enrichment | |
|---|---|---|---|---|
| Upregulated in Progressors | lipid metabolic process | 33 | 6.50 × 10−7 | 3.6 |
| cellular ketone metabolic process | 27 | 9.00 × 10−7 | 4.2 | |
| carboxylic acid metabolic process | 26 | 1.70 × 10−6 | 4.1 | |
| oxoacid metabolic process | 26 | 1.70 × 10−6 | 4.1 | |
| organic acid metabolic process | 26 | 1.30 × 10−6 | 4.1 | |
| monocarboxylic acid metabolic process | 19 | 4.00 × 10−6 | 5.4 | |
| cellular lipid metabolic process | 25 | 4.50 × 10−6 | 3.9 | |
| fatty acid metabolic process | 15 | 2.70 × 10−5 | 6.2 | |
| oxidation reduction | 25 | 1.30 × 10–3 | 2.8 | |
| lipid biosynthetic process | 15 | 3.60 × 10−3 | 4 | |
| Downregulated in Progressors | acute inflammatory response | 7 | 7.60 × 10−4 | 20.1 |
| acute-phase response | 5 | 2.50 × 10−3 | 38.8 | |
| inflammatory response | 8 | 9.70 × 10−3 | 8.3 | |
| defense response | 9 | 9.00 × 10−2 | 4.7 | |
| response to wounding | 8 | 8.10 × 10−2 | 5.4 | |
| response to external stimulus | 10 | 1.40 × 10−1 | 3.6 | |
| fat cell differentiation | 4 | 1.90 × 10−1 | 15.3 | |
| localization | 21 | 3.40 × 10−1 | 1.8 | |
| transport | 19 | 3.90 × 10−1 | 1.9 | |
| brown fat cell differentiation | 3 | 3.60 × 10−1 | 24.1 |
| Term | Gene Count | Benjamini | Fold Enrichment |
|---|---|---|---|
| Upregulated | |||
| PPAR signaling pathway | 12 | 5.80 × 10−6 | 9.1 |
| drug metabolism | 11 | 1.80 × 10−5 | 8.8 |
| fatty acid metabolism | 8 | 2.80 × 10−4 | 10.6 |
| metabolism of xenobiotics by cytochrome P450 | 9 | 3.10 × 10−4 | 8.2 |
| Retinol metabolism | 9 | 3.10 × 10−4 | 7.9 |
| valine, leucine and isoleucine degradation | 6 | 1.70 × 10−2 | 7.8 |
| glutathione metabolism | 6 | 2.50 × 10−2 | 6.9 |
| arachidonic acid metabolism | 7 | 3.30 × 10−2 | 5 |
| pyruvate metabolism | 5 | 5.50 × 10−2 | 7.3 |
| biosynthesis of unsaturated fatty acids | 4 | 1.00 × 10−1 | 8.9 |
| Downregulated | |||
| drug metabolism | 3 | 8.60 × 10−1 | 9.6 |
| cytokine-cytokine receptor interaction | 4 | 8.70 × 10−1 | 3.9 |
| Gene Symbol | Description | p-Value | Fold-Change |
|---|---|---|---|
| PPAR Signaling Pathway | |||
| Cd36 | CD36 antigen | 1.54 × 10−4 | 3.13 |
| Acaa1b | acetyl-Coenzyme A acyltransferase 1B | 2.65 × 10−5 | 1.79 |
| Acsl5 | acyl-CoA synthetase long-chain family member 5 | 1.90 × 10−4 | 1.5 |
| Cpt1b | carnitine palmitoyltransferase 1b, muscle | 5.93 × 10−3 | 1.67 |
| Cyp4a14 | cytochrome P450, family 4, subfamily a, polypeptide 14 | 2.50 × 10−6 | 3.58 |
| Ehhadh | enoyl-Coenzyme A, hydratase/3-hydroxyacyl Coenzyme A dehydrogenase | 5.70 × 10−6 | 1.95 |
| Fabp4 | fatty acid binding protein 4, adipocyte | 3.04 × 10−4 | 6.39 |
| Lpl | lipoprotein lipase; similar to Lipoprotein lipase precursor (LPL) | 3.64 × 10−5 | 6.26 |
| Pltp | phospholipid transfer protein | 6.42 × 10−5 | 2.37 |
| Me1 | malic enzyme 1, NADP(+)-dependent, cytosolic | 9.10 × 10−6 | 2.25 |
| Cyp4a31 | cytochrome P450, family 4, subfamily a, polypeptide 31; | 1.03 × 10−5 | 1.86 |
| Cyp4a10 | cytochrome P450, family 4, subfamily a, polypeptide 10; | 3.92 × 10−5 | 1.75 |
| Cyp4a32 | cytochrome P450, family 4, subfamily a, polypeptide 32; | 4.59 × 10−5 | 1.83 |
| Scd1 | stearoyl-Coenzyme A desaturase 1 | 1.11 × 10−4 | 1.51 |
| Lipid Metabolic Process | |||
| Agpat9 | 1-acylglycerol-3-phosphate O-acyltransferase 9 | 1.80 × 10−3 | 1.7 |
| Cd74 | CD74 antigen (invariant polypeptide of major histocompatibility complex, class II antigen-associated) | 2.75 × 10−3 | 1.62 |
| Elovl5 | ELOVL family member 5, elongation of long chain fatty acids (yeast) | 6.05 × 10−5 | 2.05 |
| Elovl7 | ELOVL family member 7, elongation of long chain fatty acids (yeast) | 1.12 × 10−2 | 2.1 |
| 9130409I23Rik | RIKEN cDNA 9130409I23 gene | 1.33 × 10−4 | 2.63 |
| Acot2 | acyl-CoA thioesterase 2 | 2.90 × 10−3 | 1.93 |
| Acer2 | alkaline ceramidase 2 | 1.63 × 10−3 | 1.77 |
| Crat | carnitine acetyltransferase | 1.88 × 10−3 | 1.68 |
| Cidea | cell death-inducing DNA fragmentation factor, α subunit-like effector A | 3.01 × 10−3 | 8.58 |
| Cyp17a1 | cytochrome P450, family 17, subfamily a, polypeptide 1 | 7.22 × 10−3 | 2.7 |
| Elovl3 | elongation of very long chain fatty acids (FEN1/Elo2, SUR4/Elo3, yeast)-like 3 | 3.52 × 10−4 | 1.83 |
| Ebpl | emopamil binding protein-like | 6.39 × 10−5 | 1.51 |
| Fasn | fatty acid synthase | 4.53 × 10−4 | 1.93 |
| Far2 | fatty acyl CoA reductase 2 | 3.78 × 10−3 | 1.73 |
| Osbpl3 | oxysterol binding protein-like 3 | 2.03 × 10−3 | 2.63 |
| Pctp | phosphatidylcholine transfer protein | 7.83 × 10−4 | 2.14 |
| Pik3c2g | phosphatidylinositol 3-kinase, C2 domain containing, γ polypeptide | 1.53 × 10−5 | 1.66 |
| Rbp1 | retinol binding protein 1, cellular | 9.88 × 10−5 | 2.14 |
| Rdh11 | retinol dehydrogenase 11 | 1.70 × 10−2 | 1.5 |
| Rdh16 | retinol dehydrogenase 16 | 6.55 × 10−5 | 1.97 |
| Rdh9 | retinol dehydrogenase 9 | 8.30 × 10−4 | 2.24 |
| Sigmar1 | sigma non-opioid intracellular receptor 1 | 1.40 × 10−4 | 1.51 |
| Hmgcs1 | 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 1 | 1.42 × 10−2 | 1.65 |
| Mogat1 | monoacylglycerol O-acyltransferase 1 | 2.20 × 10−4 | 2.78 |
| Vldlr | very low density lipoprotein receptor | 5.65 × 10−5 | 2.45 |
| Inflammatory Response | |||
| Cxcl1 | chemokine (C-X-C motif) ligand 1 | 1.64 × 10−2 | 0.65 |
| Mbl1 | mannose-binding lectin (protein A) 1 | 1.72 × 10−4 | 0.64 |
| Serpina3n | serine (or cysteine) peptidase inhibitor, clade A, member 3N | 4.95 × 10−4 | 0.58 |
| Saa1 | serum amyloid A 1 | 1.27 × 10−5 | 0.17 |
| Saa2 | serum amyloid A 2 | 4.27 × 10−5 | 0.17 |
| Saa4 | serum amyloid A 4 | 5.13 × 10−5 | 0.54 |
| C4a | similar to Complement C4 precursor | 5.89 × 10−4 | 0.64 |
| Stat3 | signal transducer and activator of transcription 3 | 2.60 × 10−4 | 0.65 |

3. Discussion
4. Experimental Section
4.1. Animals and Treatment
4.2. Biological Analysis and Insulin Tolerance Test
4.3. Hyperinsulinemic-Euglycemic Clamp Study
4.4. Liver Histopathology
4.5. Preparation of RNA and Microarray Hybridization
4.6. GeneChip Microarray Analysis
4.7. Quantitative Real-Time PCR
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Angulo, P.; Lindor, K.D. Non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2002, 17, S186–S190. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S.; Marino, M. Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD). Ann. Hepatol. 2009, 8, S4–S8. [Google Scholar] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.W.; Adams, L.A. Nonalcoholic fatty liver disease and diabetes mellitus: Pathogenesis and treatment. Nat. Rev. Endocrinol. 2011, 7, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Houlihan, D.D.; Rowe, I.A. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 363, 1185. [Google Scholar] [PubMed]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Fruchart, J.C. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes 2005, 54, 2460–2470. [Google Scholar] [CrossRef] [PubMed]
- Bedoucha, M.; Atzpodien, E.; Boelsterli, U.A. Diabetic KKAy mice exhibit increased hepatic PPARγ1 gene expression and develop hepatic steatosis upon chronic treatment with antidiabetic thiazolidinediones. J. Hepatol. 2001, 35, 17–23. [Google Scholar] [CrossRef]
- Rahimian, R.; Masih-Khan, E.; Lo, M.; van Breemen, C.; McManus, B.M.; Dube, G.P. Hepatic over-expression of peroxisome proliferator activated receptor γ2 in the ob/ob mouse model of non-insulin dependent diabetes mellitus. Mol. Cell. Biochem. 2001, 224, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Memon, R.A.; Tecott, L.H.; Nonogaki, K.; Beigneux, A.; Moser, A.H.; Grunfeld, C.; Feingold, K.R. Up-regulation of peroxisome proliferator-activated receptors (PPAR-α) and PPAR-γ messenger ribonucleic acid expression in the liver in murine obesity: Troglitazone induces expression of PPAR-γ-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 2000, 141, 4021–4031. [Google Scholar] [PubMed]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [PubMed]
- Moran-Salvador, E.; Lopez-Parra, M.; Garcia-Alonso, V.; Titos, E.; Martinez-Clemente, M.; Gonzalez-Periz, A.; Lopez-Vicario, C.; Barak, Y.; Arroyo, V.; Claria, J. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, I.; Rodriguez-Juan, C.; Diaz-Sanjuan, T.; Martinez, M.A.; Munoz-Yague, T.; Solis-Herruzo, J.A. Effects of rosiglitazone on the liver histology and mitochondrial function in ob/ob mice. Hepatology (Baltimore, Md.) 2007, 46, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Rull, A.; Geeraert, B.; Aragones, G.; Beltran-Debon, R.; Rodriguez-Gallego, E.; Garcia-Heredia, A.; Pedro-Botet, J.; Joven, J.; Holvoet, P.; Camps, J. Rosiglitazone and fenofibrate exacerbate liver steatosis in a mouse model of obesity and hyperlipidemia. A transcriptomic and metabolomic study. J. Proteome Res. 2014, 13, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.M.; Han, F.; Kong, L.B.; Zhao, S.X.; Wang, R.Q.; Wu, W.J.; Yu, J. Adenovirus-mediated peroxisome proliferator activated receptor γ overexpression prevents nutritional fibrotic steatohepatitis in mice. Scand. J. Gastroenterol. 2011, 46, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Chu, E.S.; Lam, C.N.; Cheng, A.S.; Lee, C.W.; Wong, V.W.; Sung, J.J.; Yu, J. PPARγ is essential for protection against nonalcoholic steatohepatitis. Gene Ther. 2010, 17, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.A.; Liu, J.Z.; Ren, Y.; Minze, L.J.; Wiles, J.R.; Collins, A.R.; Lyon, C.J.; Pratico, D.; Finegold, M.J.; Wong, S.T.; et al. Rosiglitazone attenuates age- and diet-associated nonalcoholic steatohepatitis in male low-density lipoprotein receptor knockout mice. Hepatology 2010, 52, 2001–2211. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Morais, A.; Lebrun, V.; Abarca-Quinones, J.; Brichard, S.; Hue, L.; Guigas, B.; Viollet, B.; Leclercq, I.A. Prevention of steatohepatitis by pioglitazone: Implication of adiponectin-dependent inhibition of SREBP-1c and inflammation. J. Hepatol. 2009, 50, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Huan, Y.; Jiang, Q.; Sun, S.-J.; Jia, C.-M.; Shen, Z.-F. Effects and potential mechanisms of pioglitazone on lipid metabolism in obese diabetic KKAy mice. PPAR Res. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Soejima, Y.; Fukusato, T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2300–2308. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Greco, D.; Kotronen, A.; Westerbacka, J.; Puig, O.; Arkkila, P.; Kiviluoto, T.; Laitinen, S.; Kolak, M.; Fisher, R.M.; Hamsten, A.; et al. Gene expression in human NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1281–G1287. [Google Scholar] [CrossRef] [PubMed]
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Siren, J.; Hamsten, A.; Fisher, R.M.; Yki-Jarvinen, H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007, 56, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Abu-Elheiga, L.; Matzuk, M.M.; Kordari, P.; Oh, W.; Shaikenov, T.; Gu, Z.; Wakil, S.J. Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proc. Natl. Acad. Sci. USA 2005, 102, 12011–12016. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Kim, Y.C.; Ntambi, J.M. A lipogenic diet in mice with a disruption of the stearoyl-CoA desaturase 1 gene reveals a stringent requirement of endogenous monounsaturated fatty acids for triglyceride synthesis. J. Lipid Res. 2001, 42, 1018–1024. [Google Scholar] [PubMed]
- Morgan, K.; Uyuni, A.; Nandgiri, G.; Mao, L.; Castaneda, L.; Kathirvel, E.; French, S.W.; Morgan, T.R. Altered expression of transcription factors and genes regulating lipogenesis in liver and adipose tissue of mice with high fat diet-induced obesity and nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2008, 20, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Matsusue, K.; Kashireddy, P.; Cao, W.Q.; Yeldandi, V.; Yeldandi, A.V.; Rao, M.S.; Gonzalez, F.J.; Reddy, J.K. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma 1 (PPARγ1) overexpression. J. Biol. Chem. 2003, 278, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Malle, E.; de Beer, F.C. Human serum amyloid A (SAA) protein: A prominent acute-phase reactant for clinical practice. Eur. J. Clin. Investig. 1996, 26, 427–435. [Google Scholar] [CrossRef]
- Lin, Y.; Rajala, M.W.; Berger, J.P.; Moller, D.E.; Barzilai, N.; Scherer, P.E. Hyperglycemia-induced production of acute phase reactants in adipose tissue. J. Biol. Chem. 2001, 276, 42077–42083. [Google Scholar] [CrossRef] [PubMed]
- Scheja, L.; Heese, B.; Zitzer, H.; Michael, M.D.; Siesky, A.M.; Pospisil, H.; Beisiegel, U.; Seedorf, K. Acute-phase serum amyloid A as a marker of insulin resistance in mice. Exp. Diabetes Res. 2008, 2008, 230837. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Z.; Lee, M.J.; Hu, H.; Pollin, T.I.; Ryan, A.S.; Nicklas, B.J.; Snitker, S.; Horenstein, R.B.; Hull, K.; Goldberg, N.H.; et al. Acute-phase serum amyloid A: An inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006, 3, e287. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.D.; Kip, K.E.; Marroquin, O.C.; Ridker, P.M.; Kelsey, S.F.; Shaw, L.J.; Pepine, C.J.; Sharaf, B.; Bairey Merz, C.N.; Sopko, G.; et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: The national heart, lung, and blood institute-sponsored womenʼs ischemia syndrome evaluation (WISE). Circulation 2004, 109, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, P.; Teppo, A.M.; Koistinen, H.A.; Viikari, J.; Ronnemaa, T.; Nissen, M.; Bergkulla, S.; Salmela, P.; Saltevo, J.; Koivisto, V.A. Troglitazone reduces hyperglycaemia and selectively acute-phase serum proteins in patients with Type II diabetes. Diabetologia 1999, 42, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Subramanian, S.; Chan, C.K.; Omer, M.; Chiba, T.; Wight, T.N.; Chait, A. Adipocyte-derived serum amyloid A3 and hyaluronan play a role in monocyte recruitment and adhesion. Diabetes 2007, 56, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Way, J.M.; Harrington, W.W.; Brown, K.K.; Gottschalk, W.K.; Sundseth, S.S.; Mansfield, T.A.; Ramachandran, R.K.; Willson, T.M.; Kliewer, S.A. Comprehensive messenger ribonucleic acid profiling reveals that peroxisome proliferator-activated receptor γ activation has coordinate effects on gene expression in multiple insulin-sensitive tissues. Endocrinology 2001, 142, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Burant, C.F.; Sreenan, S.; Hirano, K.; Tai, T.A.; Lohmiller, J.; Lukens, J.; Davidson, N.O.; Ross, S.; Graves, R.A. Troglitazone action is independent of adipose tissue. J. Clin. Investig. 1997, 100, 2900–2908. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Fillmore, J.J.; Gavrilova, O.; Chao, L.; Higashimori, T.; Choi, H.; Kim, H.J.; Yu, C.; Chen, Y.; Qu, X.; et al. Differential effects of rosiglitazone on skeletal muscle and liver insulin resistance in A-ZIP/F-1 fatless mice. Diabetes 2003, 52, 1311–1318. [Google Scholar]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; Olefsky, J.M.; Reichart, D.; Nguyen, M.T.; Bandyopadyhay, G.; Leung, H.Y.; Watt, M.J.; Benner, C.; Febbraio, M.A.; Nguyen, A.K.; et al. Macrophage PPAR γ is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J. Clin. Investig. 2007, 117, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Q.; Sun, S.; Jiang, Q.; Peng, J.; Shen, Z. The application of 2-NBDG as a fluorescent tracer for assessing hepatic glucose production in mice during hyperinsulinemic euglycemic clamp. Acta Pharm. Sin. B 2012, 2, 403–410. [Google Scholar] [CrossRef]
- Palmer, C.N.; Hsu, M.H.; Griffin, K.J.; Raucy, J.L.; Johnson, E.F. Peroxisome proliferator activated receptor-α expression in human liver. Mol. Pharmacol. 1998, 53, 14–22. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, C.; Huan, Y.; Liu, S.; Hou, S.; Sun, S.; Li, C.; Liu, Q.; Jiang, Q.; Wang, Y.; Shen, Z. Effect of Chronic Pioglitazone Treatment on Hepatic Gene Expression Profile in Obese C57BL/6J Mice. Int. J. Mol. Sci. 2015, 16, 12213-12229. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160612213
Jia C, Huan Y, Liu S, Hou S, Sun S, Li C, Liu Q, Jiang Q, Wang Y, Shen Z. Effect of Chronic Pioglitazone Treatment on Hepatic Gene Expression Profile in Obese C57BL/6J Mice. International Journal of Molecular Sciences. 2015; 16(6):12213-12229. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160612213
Chicago/Turabian StyleJia, Chunming, Yi Huan, Shuainan Liu, Shaocong Hou, Sujuan Sun, Caina Li, Quan Liu, Qian Jiang, Yue Wang, and Zhufang Shen. 2015. "Effect of Chronic Pioglitazone Treatment on Hepatic Gene Expression Profile in Obese C57BL/6J Mice" International Journal of Molecular Sciences 16, no. 6: 12213-12229. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160612213