
Pathophysiology of Non Alcoholic Fatty Liver Disease

 , ,
, ,  , and
, and 
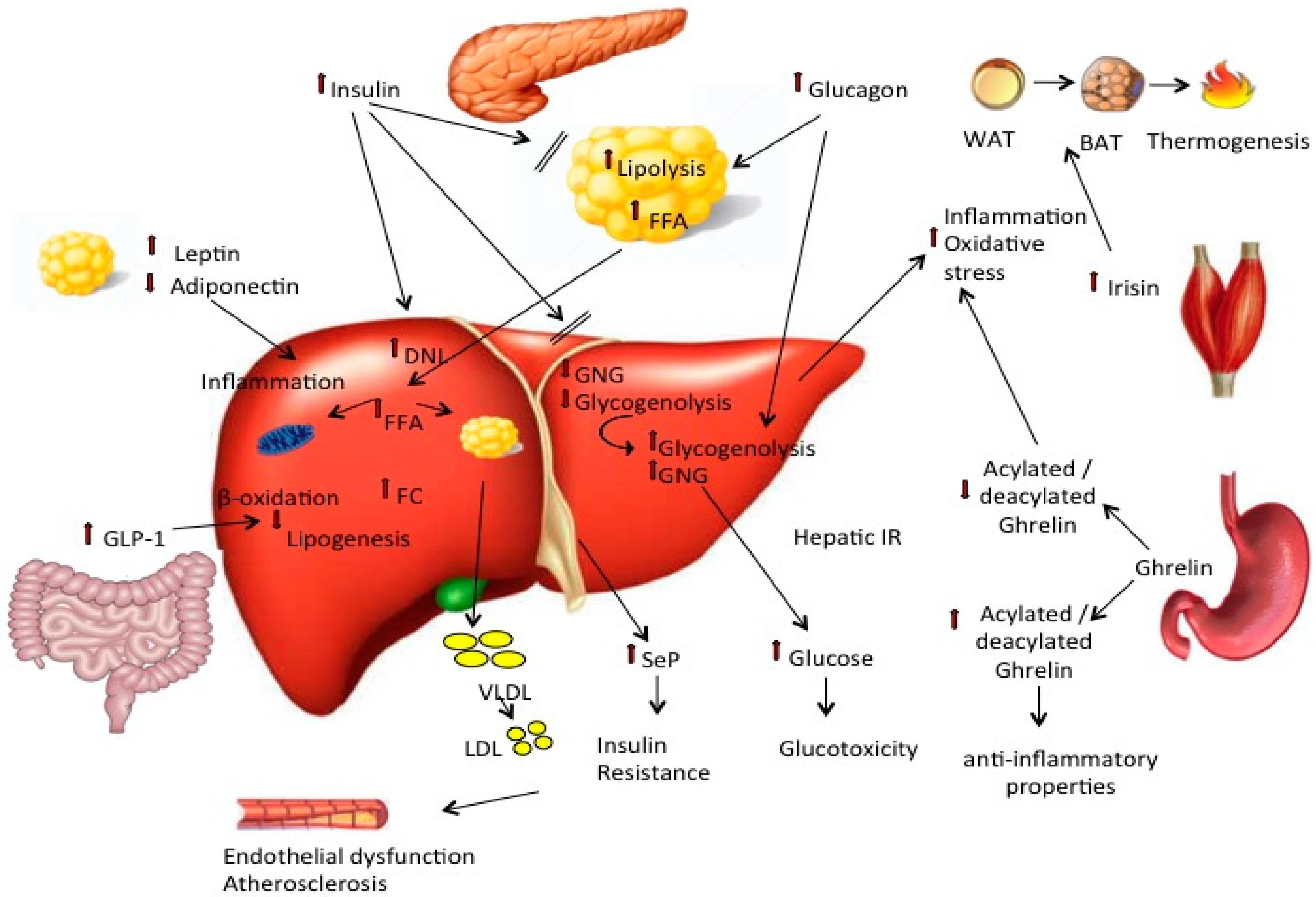{kind=link}
Abstract
:1. Introduction
2. Circulating Lipids
2.1. Free Fatty Acids (FFA)
2.1.1. FFA and Diet
2.1.2. FFA and Exercise
2.1.3. FFA and Inflammation
2.1.4. FFA and Nonalcoholic Fatty Liver Disease (NAFLD)
2.1.5. FFA and Kidney
2.2. Cholesterol
2.2.1. Cholesterol and Diet
2.2.2. Cholesterol and Exercise
2.2.3. Cholesterol and Inflammation
2.2.4. Cholesterol and NAFLD
2.2.5. Cholesterol and Kidney
3. Adipose Tissue Released Compounds
3.1. Adiponectin
3.1.1. Adiponectin and Diet
3.1.2. Adiponectin and Exercise
3.1.3. Adiponectin and Inflammation
3.1.4. Adiponectin and NAFLD
3.1.5. Adiponectin and Kidney
3.2. Leptin
3.2.1. Leptin and Diet
3.2.2. Leptin and Exercise
3.2.3. Leptin and Inflammation
3.2.4. Leptin and NAFLD
3.2.5. Leptin and Kidney
4. Pancreatic Hormones and NAFLD
4.1. Insulin
4.1.1. Insulin and Diet
4.1.2. Insulin and NAFLD
4.1.3. Insulin and Exercise
4.2. Glucagon
4.2.1. Glucagon and Diet
4.2.2. Glucagon and NAFLD
4.2.3. Glucagon and Exercise
4.2.4. Glucagon and Inflammation
5. Gut Released Hormones
5.1. GLP-1
5.1.1. GLP-1 and Diet
5.1.2. GLP-1 and Exercise
5.1.3. GLP-1 and Inflammation
5.1.4. GLP-1 and NAFLD
5.1.5. GLP-1 and Kidney
5.2. Ghrelin
5.2.1. Ghrelin and Diet
5.2.2. Ghrelin and Exercise
5.2.3. Ghrelin and Inflammation
5.2.4. Ghrelin and NAFLD
5.2.5. Ghrelin and Kidney
6. Muscle Released Compounds
6.1. Irisin
6.1.1. Irisin and Diet
6.1.2. Irisin and Exercise
6.1.3. Irisin, NAFLD and Inflammation
6.1.4. Irisin and Kidney
7. Liver-Released Compounds
7.1. Selenoprotein P
7.1.1. Selenoprotein P and Diet
7.1.2. Selenoprotein P and Exercise
7.1.3. Selenoprotein P and Inflammation
7.1.4. Selenoprotein P and NAFLD
7.1.5. Selenoprotein P and Kidney
7.2. Fetuin-A
7.2.1. Fetuin-A and Diet
7.2.2. Fetuin-A and Exercise
7.2.3. Fetuin-A and Inflammation
7.2.4. Fetuin-A and NAFLD
7.2.5. Fetuin-A and Kidney
8. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| AG | acylated ghrelin |
| AMPK | adenosine monophosphate-activated protein kinase |
| ANP | atrial natriuretic peptide |
| BAT | brown adipose tissue |
| BMI | body mass index |
| CIMT | carotid intima-media thickness |
| CKD | chronic kidney disease |
| CRP | C-reactive protein |
| DAG | diacyl glycerol |
| DeAG | des-acylated ghrelin |
| DKD | diabetic kidney disease |
| DNL | de novo lipogenesis |
| ELISA | enzyme-linked immunosorbent assay |
| FC | free cholesterol |
| FFA | free fatty acid |
| FNDC5 | fibronectin type III domain-containing protein 5 |
| FoxO3a | forkhead box O3a |
| FSGS | focal segmental glomerular sclerosis |
| GLP-1 | glucagon-like peptide 1 |
| GLP-1R | glucagon-like peptide 1 receptor |
| GNG | gluconeogenesis |
| GOAT | ghrelin-ghrelin O-acyltransferase |
| (Oct)-1 | organic cation transporter |
| IR | insulin resistance |
| IS | insulin sensitive |
| JNK | c-Jun terminal kinase |
| LDL | low density lipoprotein |
| MRS | magnetic resonance spectroscopy |
| MS | metabolic syndrome |
| MUFA | monounsaturated fatty acids |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGT | normal glucose tolerance |
| NLRP3 | nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 |
| NO | nitric oxide |
| OGTT | oral glucose tolerance test |
| PUFA | polyunsaturated fatty acids |
| SC | subcutaneous |
| Se | selenium |
| SeP | selenoprotein P |
| SEPP1 | selenoprotein P, plasma 1 |
| SFA | saturated fatty acids |
| T2D | type 2 diabetes |
| TLR | toll like receptors |
| TNF-α | tumor necrosis factor-α |
| UCP1 | uncoupling protein 1 |
| VS | visceral |
| VLDL | very low density lipoprotein |
| WAT | white adipose tissue |
References
- Groop, L.C.; Bonadonna, R.C.; DelPrato, S.; Ratheiser, K.; Zyck, K.; Ferrannini, E.; DeFronzo, R.A. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J. Clin. Investig. 1989, 84, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Lafontan, M.; Langin, D. Lipolysis and lipid mobilization in human adipose tissue. Prog. Lipid Res. 2009, 48, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Camastra, S.; Coppack, S.W.; Fliser, D.; Golay, A.; Mitrakou, A. Insulin action and non-esterified fatty acids. The European Group for the Study of Insulin Resistance (EGIR). Proc. Nutr. Soc. 1997, 56, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Legrand-Poels, S.; Esser, N.; L’Homme, L.; Scheen, A.; Paquot, N.; Piette, J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem. Pharmacol. 2014, 92, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.P.; Texeira, T.F.; Ferreira, A.B.; Peluzio Mdo, C.; Alfenas Rde, C. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br. J. Nutr. 2012, 108, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Mittendorfer, B.; Yoshino, M.; Patterson, B.W.; Klein, S. VLDL triglyceride kinetics in lean, overweight, and obese men and women. J. Clin. Endocrinol. Metab. 2016, 101, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.J.; Mente, A.; Maroleanu, A.; Cozma, A.I.; Ha, V.; Kishibe, T.; Uleryk, E.; Budylowski, P.; Schunemann, H.; Beyene, J.; et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: Systematic review and meta-analysis of observational studies. BMJ 2015, 351, h3978. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated Fatty acids. J. Nutr. Metab. 2012, 2012, 539426. [Google Scholar] [CrossRef] [PubMed]
- Maehre, H.K.; Jensen, I.J.; Elvevoll, E.O.; Eilertsen, K.E. Omega-3 fatty acids and cardiovascular diseases: Effects, mechanisms and dietary relevance. Int. J. Mol. Sci. 2015, 16, 22636–22661. [Google Scholar] [CrossRef] [PubMed]
- Romijn, J.A.; Coyle, E.F.; Sidossis, L.S.; Gastaldelli, A.; Horowitz, J.F.; Endert, E.; Wolfe, R.R. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am. J. Physiol. 1993, 265, E380–E391. [Google Scholar] [PubMed]
- Boden, G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Curr. Diabetes Rep. 2006, 6, 177–181. [Google Scholar] [CrossRef]
- Gadang, V.; Kohli, R.; Myronovych, A.; Hui, D.Y.; Perez-Tilve, D.; Jaeschke, A. MLK3 promotes metabolic dysfunction induced by saturated fatty acid-enriched diet. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E549–E556. [Google Scholar] [CrossRef] [PubMed]
- Leamy, A.K.; Egnatchik, R.A.; Shiota, M.; Ivanova, P.T.; Myers, D.S.; Brown, H.A.; Young, J.D. Enhanced synthesis of saturated phospholipids is associated with ER stress and lipotoxicity in palmitate treated hepatic cells. J. Lipid Res. 2014, 55, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Snodgrass, R.G.; Huang, S.; Choi, I.W.; Rutledge, J.C.; Hwang, D.H. Inflammasome-mediated secretion of IL-1β in human monocytes through TLR2 activation; modulation by dietary fatty acids. J. Immunol. 2013, 191, 4337–4347. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Mondal, A.K.; Elbein, S.C. Distinct gene expression profiles characterize cellular responses to palmitate and oleate. J. Lipid Res. 2010, 51, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Badeanlou, L.; Bielawski, J.; Roberts, A.J.; Hannun, Y.A.; Samad, F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E211–E224. [Google Scholar] [CrossRef] [PubMed]
- Ussher, J.R.; Koves, T.R.; Cadete, V.J.; Zhang, L.; Jaswal, J.S.; Swyrd, S.J.; Lopaschuk, D.G.; Proctor, S.D.; Keung, W.; Muoio, D.M.; et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes 2010, 59, 2453–2464. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E275–E281. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Zhou, Y.; Sadevirta, S.; Leivonen, M.; Arola, J.; Oresic, M.; Hyotylainen, T.; Yki-Jarvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Yang, L.; Fabbrini, E.; Mohammed, B.S.; Eagon, J.C.; Hotamisligil, G.S.; Klein, S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 2009, 58, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Mantzaris, M.D.; Tsianos, E.V.; Galaris, D. Interruption of triacylglycerol synthesis in the endoplasmic reticulum is the initiating event for saturated fatty acid-induced lipotoxicity in liver cells. FEBS J. 2011, 278, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The subtle balance between lipolysis and lipogenesis: A critical point in metabolic homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Sieber, J.; Jehle, A.W. Free fatty acids and their metabolism affect function and survival of podocytes. Front. Endocrinol. 2014, 5, 186. [Google Scholar] [CrossRef] [PubMed]
- Lennon, R.; Pons, D.; Sabin, M.A.; Wei, C.; Shield, J.P.; Coward, R.J.; Tavare, J.M.; Mathieson, P.W.; Saleem, M.A.; Welsh, G.I. Saturated fatty acids induce insulin resistance in human podocytes: Implications for diabetic nephropathy. Nephrol. Dial. Transplant. 2009, 24, 3288–3296. [Google Scholar] [CrossRef] [PubMed]
- Sieber, J.; Lindenmeyer, M.T.; Kampe, K.; Campbell, K.N.; Cohen, C.D.; Hopfer, H.; Mundel, P.; Jehle, A.W. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am. J. Physiol. Ren. Physiol. 2010, 299, F821–F829. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Janoudi, A.; Shamoun, F.E.; Kalavakunta, J.K.; Abela, G.S. Cholesterol crystal induced arterial inflammation and destabilization of atherosclerotic plaque. Eur. Heart J. 2016, 37, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Keys, A. Coronary heart disease, serum cholesterol, and the diet. Acta Med. Scand. 1980, 207, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Franklin, B.A.; Durstine, J.L.; Roberts, C.K.; Barnard, R.J. Impact of diet and exercise on lipid management in the modern era. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, J.K.; Mursu, J.; Virtanen, H.E.; Fogelholm, M.; Salonen, J.T.; Koskinen, T.T.; Voutilainen, S.; Tuomainen, T.P. Associations of egg and cholesterol intakes with carotid intima-media thickness and risk of incident coronary artery disease according to apolipoprotein E phenotype in men: The Kuopio Ischaemic Heart Disease Risk Factor Study. Am. J. Clin. Nutr. 2016, 103, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Caesar, R.; Nygren, H.; Oresic, M.; Backhed, F. Interaction between dietary lipids and gut microbiota regulates hepatic cholesterol metabolism. J. Lipid Res. 2016, 57, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Mann, S.; Beedie, C.; Jimenez, A. Differential effects of aerobic exercise, resistance training and combined exercise modalities on cholesterol and the lipid profile: Review, synthesis and recommendations. Sports Med. 2014, 44, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donovan, G.; Owen, A.; Bird, S.R.; Kearney, E.M.; Nevill, A.M.; Jones, D.W.; Woolf-May, K. Changes in cardiorespiratory fitness and coronary heart disease risk factors following 24 wk of moderate- or high-intensity exercise of equal energy cost. J. Appl. Physiol. 2005, 98, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Lira, F.S.; Yamashita, A.S.; Uchida, M.C.; Zanchi, N.E.; Gualano, B.; Martins, E., Jr.; Caperuto, E.C.; Seelaender, M. Low and moderate, rather than high intensity strength exercise induces benefit regarding plasma lipid profile. Diabetol. Metab. Syndr. 2010, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Katzmarzyk, P.T.; Leon, A.S.; Rankinen, T.; Gagnon, J.; Skinner, J.S.; Wilmore, J.H.; Rao, D.C.; Bouchard, C. Changes in blood lipids consequent to aerobic exercise training related to changes in body fatness and aerobic fitness. Metabolism 2001, 50, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Schwabe, R.F.; Devries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6: Model of NF-κB and map kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Min, H.K.; Kapoor, A.; Fuchs, M.; Mirshahi, F.; Zhou, H.; Maher, J.; Kellum, J.; Warnick, R.; Contos, M.J.; Sanyal, A.J. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012, 15, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Wahl, P.; Ducasa, G.M.; Fornoni, A. Systemic and renal lipids in kidney disease development and progression. Am. J. Physiol. Ren. Physiol. 2016, 310, F433–F445. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Kruth, H.S. Accumulation of cholesterol in the lesions of focal segmental glomerulosclerosis. Nephrology 2003, 8, 224–223. [Google Scholar] [CrossRef] [PubMed]
- Fornoni, A.; Merscher, S.; Kopp, J.B. Lipid biology of the podocyte—New perspectives offer new opportunities. Nat. Rev. Nephrol. 2014, 10, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Jiang, T.; Shen, Y.; Caldas, Y.; Miyazaki-Anzai, S.; Santamaria, H.; Urbanek, C.; Solis, N.; Scherzer, P.; Lewis, L.; et al. Diabetic nephropathy is accelerated by farnesoid X receptor deficiency and inhibited by farnesoid X receptor activation in a type 1 diabetes model. Diabetes 2010, 59, 2916–2927. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Smith, M.W.; Nelson, G.W.; Johnson, R.C.; Freedman, B.I.; Bowden, D.W.; Oleksyk, T.; McKenzie, L.M.; Kajiyama, H.; Ahuja, T.S.; et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat. Genet. 2008, 40, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.; Kranzlin, B.; Wagenblabeta, K.; Bonrouhi, M.; Thiery, J.; Grone, E.; Nordstrom, V.; Teupser, D.; Gretz, N.; Malle, E.; et al. Lipid droplet accumulation is associated with an increase in hyperglycemia-induced renal damage: Prevention by liver X receptors. Am. J. Pathol. 2013, 182, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R. Effects of statins on renal function. Am. J. Cardiol. 2006, 97, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Scudiero, O.; Monaco, M.L.; Palmieri, A.; Mazzarella, G.; Costagliola, C.; Bianco, A.; Daniele, A. New insight into adiponectin role in obesity and obesity-related diseases. BioMed Res. Int. 2014, 2014, 658913. [Google Scholar] [CrossRef] [PubMed]
- Scherer, P.E. The multifaceted roles of adipose tissue-therapeutic targets for diabetes and beyond: The 2015 banting lecture. Diabetes 2016, 65, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Freitas Lima, L.C.; Braga, V.A.; do Socorro de Franca Silva, M.; Cruz, J.C.; Sousa Santos, S.H.; de Oliveira Monteiro, M.M.; Balarini, C.M. Adipokines, diabetes and atherosclerosis: An inflammatory association. Front. Physiol. 2015, 6, 304. [Google Scholar] [CrossRef] [PubMed]
- Von Frankenberg, A.D.; Silva, F.M.; de Almeida, J.C.; Piccoli, V.; do Nascimento, F.V.; Sost, M.M.; Leitao, C.B.; Remonti, L.L.; Umpierre, D.; Reis, A.F.; et al. Effect of dietary lipids on circulating adiponectin: A systematic review with meta-analysis of randomised controlled trials. Br. J. Nutr. 2014, 112, 1235–1250. [Google Scholar] [CrossRef] [PubMed]
- Kamari, Y.; Grossman, E.; Oron-Herman, M.; Peleg, E.; Shabtay, Z.; Shamiss, A.; Sharabi, Y. Metabolic stress with a high carbohydrate diet increases adiponectin levels. Horm. Metab. Res. 2007, 39, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, R.; Cianflone, K.; McGahan, J.P.; Berglund, L.; Bremer, A.A.; Keim, N.L.; Griffen, S.C.; Havel, P.J.; Stanhope, K.L. Effects of sugar-sweetened beverages on plasma acylation stimulating protein, leptin and adiponectin: Relationships with metabolic outcomes. Obesity 2013, 21, 2471–2480. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, M.M.; Havel, P.J. Physiological, pharmacological, and nutritional regulation of circulating adiponectin concentrations in humans. Metab. Syndr. Relat. Disord. 2008, 6, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Golbidi, S.; Laher, I. Exercise induced adipokine changes and the metabolic syndrome. J. Diabetes Res. 2014, 2014, 726861. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, N.; Eshaghian, S.; Huizenga, R.; Sosnin, K.; Ebrahimi, R.; Siegel, R. Effects of intense exercise and moderate caloric restriction on cardiovascular risk factors and inflammation. Am. J. Med. 2011, 124, 978–982. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G. Adiponectin and inflammation: Consensus and controversy. J. Allergy Clin. Immunol. 2008, 121, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Pagotto, U.; Manini, R.; Vanni, E.; Gastaldelli, A.; de Iasio, R.; Gentilcore, E.; Natale, S.; Cassader, M.; Rizzetto, M.; et al. Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity. J. Clin. Endocrinol. Metab. 2005, 90, 3498–3504. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Harrison, S.; Belfort-Aguiar, R.; Hardies, J.; Balas, B.; Schenker, S.; Cusi, K. Pioglitazone in the treatment of NASH: The role of adiponectin. Aliment. Pharmacol. Ther. 2010, 32, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Yamauchi, T. Adiponectin and adiponectin receptors. Endocr. Rev. 2005, 26, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Hoshide, S.; Ishikawa, J.; Hashimoto, T.; Eguchi, K.; Shimada, K.; Kario, K. Differential impacts of adiponectin on low-grade albuminuria between obese and nonobese persons without diabetes. J. Clin. Hypertens. 2007, 9, 775–782. [Google Scholar] [CrossRef]
- Ohashi, K.; Iwatani, H.; Kihara, S.; Nakagawa, Y.; Komura, N.; Fujita, K.; Maeda, N.; Nishida, M.; Katsube, F.; Shimomura, I.; et al. Exacerbation of albuminuria and renal fibrosis in subtotal renal ablation model of adiponectin-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Ramachandrarao, S.; Qiu, G.; Usui, H.K.; Zhu, Y.; Dunn, S.R.; Ouedraogo, R.; Hough, K.; McCue, P.; Chan, L.; et al. Adiponectin regulates albuminuria and podocyte function in mice. J. Clin. Investig. 2008, 118, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Iwashima, Y.; Horio, T.; Kumada, M.; Suzuki, Y.; Kihara, S.; Rakugi, H.; Kawano, Y.; Funahashi, T.; Ogihara, T. Adiponectin and renal function, and implication as a risk of cardiovascular disease. Am. J. Cardiol. 2006, 98, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Ignacy, W.; Chudek, J.; Adamczak, M.; Funahashi, T.; Matsuzawa, Y.; Kokot, F.; Wiecek, A. Reciprocal association of plasma adiponectin and serum C-reactive protein concentration in haemodialysis patients with end-stage kidney disease—A follow-up study. Nephron Clin. Pract. 2005, 101, c18–c24. [Google Scholar] [CrossRef] [PubMed]
- Marchlewska, A.; Stenvinkel, P.; Lindholm, B.; Danielsson, A.; Pecoits-Filho, R.; Lonnqvist, F.; Schalling, M.; Heimburger, O.; Nordfors, L. Reduced gene expression of adiponectin in fat tissue from patients with end-stage renal disease. Kidney Int. 2004, 66, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Izadi, V.; Saraf-Bank, S.; Azadbakht, L. Dietary intakes and leptin concentrations. ARYA Atheroscler. 2014, 10, 266–272. [Google Scholar] [PubMed]
- Gastaldelli, A.; Sironi, A.M.; Ciociaro, D.; Positano, V.; Buzzigoli, E.; Giannessi, D.; Lombardi, M.; Mari, A.; Ferrannini, E. Visceral fat and beta cell function in non-diabetic humans. Diabetologia 2005, 48, 2090–2096. [Google Scholar] [CrossRef] [PubMed]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Adya, R.; Tan, B.K.; Randeva, H.S. Differential effects of leptin and adiponectin in endothelial angiogenesis. J. Diabetes Res. 2015, 2015, 648239. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Siliart, B.; Lutz, T.A.; Biourge, V.; Nguyen, P.; Dumon, H.J. Postprandial response of plasma insulin, amylin and acylated ghrelin to various test meals in lean and obese cats. Br. J. Nutr. 2010, 103, 1610–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.E.; Harmancey, R.; Stec, D.E. Lean heart: Role of leptin in cardiac hypertrophy and metabolism. World J. Cardiol. 2015, 7, 511–524. [Google Scholar] [PubMed]
- Masquio, D.C.; de Piano, A.; Sanches, P.L.; Corgosinho, F.C.; Campos, R.M.; Carnier, J.; da Silva, P.L.; Caranti, D.A.; Tock, L.; Oyama, L.M.; et al. The effect of weight loss magnitude on pro-/anti-inflammatory adipokines and carotid intima-media thickness in obese adolescents engaged in interdisciplinary weight loss therapy. Clin. Endocrinol. 2013, 79, 55–64. [Google Scholar] [CrossRef] [PubMed]
- La Cava, A.; Matarese, G. The weight of leptin in immunity. Nat. Rev. Immunol. 2004, 4, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Cumin, F.; Baum, H.P.; de Gasparo, M.; Levens, N. Removal of endogenous leptin from the circulation by the kidney. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Lindholm, B.; Lonnqvist, F.; Katzarski, K.; Heimburger, O. Increases in serum leptin levels during peritoneal dialysis are associated with inflammation and a decrease in lean body mass. J. Am. Soc. Nephrol. 2000, 11, 1303–1309. [Google Scholar] [PubMed]
- Polyzos, S.A.; Aronis, K.N.; Kountouras, J.; Raptis, D.D.; Vasiloglou, M.F.; Mantzoros, C.S. Circulating leptin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Diabetologia 2016, 59, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Mak, R.H.; Cheung, W.; Cone, R.D.; Marks, D.L. Leptin and inflammation-associated cachexia in chronic kidney disease. Kidney Int. 2006, 69, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Nakashima, A.; Qureshi, A.R.; Lindholm, B.; Heimburger, O.; Barany, P.; Stenvinkel, P. Protein-energy wasting modifies the association of ghrelin with inflammation, leptin, and mortality in hemodialysis patients. Kidney Int. 2011, 79, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E.; et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Baldi, S.; Pettiti, M.; Toschi, E.; Camastra, S.; Natali, A.; Landau, B.R.; Ferrannini, E. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: A quantitative study. Diabetes 2000, 49, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Khodabandehloo, H.; Gorgani-Firuzjaee, S.; Panahi, G.; Meshkani, R. Molecular and cellular mechanisms linking inflammation to insulin resistance and β-cell dysfunction. Transl. Res. 2016, 167, 228–256. [Google Scholar] [CrossRef] [PubMed]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; de Bittencourt, P.I., Jr.; Newsholme, P. Molecular events linking oxidative stress and inflammation to insulin resistance and β-cell dysfunction. Oxid. Med. Cell. Longev. 2015, 2015, 181643. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, R. Mechanisms linking inflammation to insulin resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, O.; Pacini, G.; Tura, A.; Holst, J.J.; Deacon, C.F.; Ahren, B. Incretin effect after oral amino acid ingestion in humans. J. Clin. Endocrinol. Metab. 2015, 100, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, R.L.; Szczepaniak, L.S.; Myhill, J.; Tamura, Y.; Uchino, H.; Giacca, A.; McGarry, J.D. The composition of dietary fat directly influences glucose-stimulated insulin secretion in rats. Diabetes 2002, 51, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, S.; Belfort, R.; Gastaldelli, A.; Pratipanawatr, T.; Berria, R.; Pratipanawatr, W.; Bajaj, M.; Mandarino, L.; DeFronzo, R.; Cusi, K. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes 2003, 52, 2461–2474. [Google Scholar] [CrossRef] [PubMed]
- Hyotylainen, T.; Jerby, L.; Petaja, E.M.; Mattila, I.; Jantti, S.; Auvinen, P.; Gastaldelli, A.; Yki-Jarvinen, H.; Ruppin, E.; Oresic, M. Genome-scale study reveals reduced metabolic adaptability in patients with non-alcoholic fatty liver disease. Nat. Commun. 2016, 7, 8994. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Perlemuter, G.; Bonvoust, F.; Dargere, D.; Parfait, B.; Vidaud, M.; Conti, M.; Huet, S.; Ba, N.; Buffet, C.; et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 2001, 34, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Ridolfi, F.; Di Sario, A.; Casini, A.; Marucci, L.; Gaggiotti, G.; Orlandoni, P.; Macarri, G.; Perego, L.; Benedetti, A.; et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: Differential effects on signal transduction pathways. Hepatology 1999, 29, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Coggan, A.R.; Raguso, C.A.; Gastaldelli, A.; Williams, B.D.; Wolfe, R.R. Regulation of glucose production during exercise at 80% of VO2 peak in untrained humans. Am. J. Physiol. 1997, 273, E348–E354. [Google Scholar] [PubMed]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef] [PubMed]
- Wewer Albrechtsen, N.J.; Kuhre, R.E.; Pedersen, J.; Knop, F.K.; Holst, J.J. The biology of glucagon and the consequences of hyperglucagonemia. Biomark. Med. 2016, 10, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. Glucagon physiology and pathophysiology in the light of new advances. Diabetologia 1985, 28, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Gromada, J.; Franklin, I.; Wollheim, C.B. α-Cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr. Rev. 2007, 28, 84–116. [Google Scholar] [CrossRef] [PubMed]
- Mitrakou, A.; Ryan, C.; Veneman, T.; Mokan, M.; Jenssen, T.; Kiss, I.; Durrant, J.; Cryer, P.; Gerich, J. Hierarchy of glycemic thresholds for counterregulatory hormone secretion, symptoms, and cerebral dysfunction. Am. J. Physiol. 1991, 260, E67–E74. [Google Scholar] [PubMed]
- Bagger, J.I.; Knop, F.K.; Lund, A.; Holst, J.J.; Vilsboll, T. Glucagon responses to increasing oral loads of glucose and corresponding isoglycaemic intravenous glucose infusions in patients with type 2 diabetes and healthy individuals. Diabetologia 2014, 57, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Rocha, D.M.; Faloona, G.R.; Unger, R.H. Glucagon-stimulating activity of 20 amino acids in dogs. J. Clin. Investig. 1972, 51, 2346–2351. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.P.; Benson, J.W.; Walter, R.M.; Ensinck, J.W. Arginine-stimulated acute phase of insulin and glucagon secretion in diabetic subjects. J. Clin. Investig. 1976, 58, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Radulescu, A.; Gannon, M.C.; Nuttall, F.Q. The effect on glucagon, glucagon-like peptide-1, total and acyl-ghrelin of dietary fats ingested with and without potato. J. Clin. Endocrinol. Metab. 2010, 95, 3385–3391. [Google Scholar] [CrossRef] [PubMed]
- Hippen, A.R. Glucagon as a potential therapy for ketosis and fatty liver. Vet. Clin. N. Am. Food Anim. Pract. 2000, 16, 267–282. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, B.B. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E671–E678. [Google Scholar] [CrossRef] [PubMed]
- Conarello, S.L.; Jiang, G.; Mu, J.; Li, Z.; Woods, J.; Zycband, E.; Ronan, J.; Liu, F.; Roy, R.S.; Zhu, L.; et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 2007, 50, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Junker, A.E.; Gluud, L.; Holst, J.J.; Knop, F.K.; Vilsboll, T. Diabetic and nondiabetic patients with nonalcoholic fatty liver disease have an impaired incretin effect and fasting hyperglucagonaemia. J. Intern. Med. 2016, 279, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Osborne, M.C.; Monia, B.P.; Bhanot, S.; Gaarde, W.A.; Reed, C.; She, P.; Jetton, T.L.; Demarest, K.T. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 2004, 53, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.J.; Moreno-Navarrete, J.M.; Sabater, M.; Ricart, W.; Fruhbeck, G.; Fernandez-Real, J.M. Circulating glucagon is associated with inflammatory mediators in metabolically compromised subjects. Eur. J. Endocrinol. 2011, 165, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Ghanim, H.; Abuaysheh, S.; Green, K.; Batra, M.; Dhindsa, S.; Makdissi, A.; Patel, R.; Chaudhuri, A. Decreased insulin secretion and incretin concentrations and increased glucagon concentrations after a high-fat meal when compared with a high-fruit and -fiber meal. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E185–E191. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Steensberg, A.; Schjerling, P. Exercise and interleukin-6. Curr. Opin. Hematol. 2001, 8, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Maachi, M.; Van Nhieu, J.T.; Jardel, C.; Bruckert, E.; Grimaldi, A.; Robert, J.J.; Capeau, J.; Hainque, B. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J. Clin. Endocrinol. Metab. 2002, 87, 2084–2089. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. Interleukin 6 in autoimmune and inflammatory diseases: A personal memoir. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Tweedell, A.; Mulligan, K.X.; Martel, J.E.; Chueh, F.Y.; Santomango, T.; McGuinness, O.P. Metabolic response to endotoxin in vivo in the conscious mouse: Role of interleukin-6. Metabolism 2011, 60, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. Enteroendocrine secretion of gut hormones in diabetes, obesity and after bariatric surgery. Curr. Opin. Pharmacol. 2013, 13, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Fava, S. Glucagon-like peptide 1 and the cardiovascular system. Curr. Diabetes Rev. 2014, 10, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Ohneda, A.; Valverde, I.; Eisentraut, A.M.; Exton, J. Characterization of the responses of circulating glucagon-like immunoreactivity to intraduodenal and intravenous administration of glucose. J. Clin. Investig. 1968, 47, 48–65. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Bowes, A.J.; Werstuck, G.H. Diabetes, hyperglycemia and accelerated atherosclerosis: Evidence supporting a role for endoplasmic reticulum (ER) stress signaling. Cardiovasc. Hematol. Disord. Drug Targets 2010, 10, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Yoo, J.H.; So, Y.S. Effect of the low- versus high-intensity exercise training on endoplasmic reticulum stress and GLP-1 in adolescents with type 2 diabetes mellitus. J. Phys. Ther. Sci. 2015, 27, 3063–3068. [Google Scholar] [CrossRef] [PubMed]
- Kodera, R.; Shikata, K.; Kataoka, H.U.; Takatsuka, T.; Miyamoto, S.; Sasaki, M.; Kajitani, N.; Nishishita, S.; Sarai, K.; Hirota, D.; et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia 2011, 54, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, M.; Mita, T.; Azuma, K.; Ebato, C.; Goto, H.; Nomiyama, T.; Fujitani, Y.; Hirose, T.; Kawamori, R.; Watada, H. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 2010, 59, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Varanasi, A.; Patel, P.; Makdissi, A.; Dhindsa, S.; Chaudhuri, A.; Dandona, P. Clinical use of liraglutide in type 2 diabetes and its effects on cardiovascular risk factors. Endocr. Pract. 2012, 18, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Lehrskov-Schmidt, L.; Lehrskov-Schmidt, L.; Nielsen, S.T.; Holst, J.J.; Moller, K.; Solomon, T.P. The effects of TNF-α on GLP-1-stimulated plasma glucose kinetics. J. Clin. Endocrinol. Metab. 2015, 100, E616–E622. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; de Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klonoff, D.C.; Buse, J.B.; Nielsen, L.L.; Guan, X.; Bowlus, C.L.; Holcombe, J.H.; Wintle, M.E.; Maggs, D.G. Exenatide effects on diabetes, obesity, cardiovascular risk factors and hepatic biomarkers in patients with type 2 diabetes treated for at least 3 years. Curr. Med. Res. Opin. 2008, 24, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, D.J.; Irwin, A.; Gardner, C.J.; Daousi, C.; Purewal, T.; Furlong, N.; Goenka, N.; Thomas, E.L.; Adams, V.L.; Pushpakom, S.P.; et al. Improved glycaemia correlates with liver fat reduction in obese, type 2 diabetes, patients given glucagon-like peptide-1 (GLP-1) receptor agonists. PLoS ONE 2012, 7, e50117. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.R.; Brady, D.E.; Torres, D.M.; Ragozzino, L.; Chalasani, N.; Harrison, S.A. Exenatide in the treatment of diabetic patients with non-alcoholic steatohepatitis: A case series. Am. J. Gastroenterol. 2010, 105, 2707–2709. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; LEAN trial team; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2015, 387, 679–690. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Marchesini, G. Time for Glucagon like peptide-1 receptor agonists treatment for patients with NAFLD? J. Hepatol. 2016, 64, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Jendle, J.; Nauck, M.A.; Matthews, D.R.; Frid, A.; Hermansen, K.; During, M.; Zdravkovic, M.; Strauss, B.J.; Garber, A.J.; LEAD-2 and LEAD-3 Study Groups. Weight loss with liraglutide, a once-daily human glucagon-like peptide-1 analogue for type 2 diabetes treatment as monotherapy or added to metformin, is primarily as a result of a reduction in fat tissue. Diabetes Obes. Metab. 2009, 11, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Buscher, A.K.; Buscher, R.; Hauffa, B.P.; Hoyer, P.F. Alterations in appetite-regulating hormones influence protein-energy wasting in pediatric patients with chronic kidney disease. Pediatr. Nephrol. 2010, 25, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.R.; Fan, X.M. Ghrelin-ghrelin O-acyltransferase system in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2015, 21, 3214–3222. [Google Scholar] [PubMed]
- Al Massadi, O.; Tschop, M.H.; Tong, J. Ghrelin acylation and metabolic control. Peptides 2011, 32, 2301–2308. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.E.; Frayo, R.S.; Marmonier, C.; Aubert, R.; Chapelot, D. Plasma ghrelin levels and hunger scores in humans initiating meals voluntarily without time- and food-related cues. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E297–E304. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A. Novel molecular aspects of ghrelin and leptin in the control of adipobiology and the cardiovascular system. Obes. Facts 2014, 7, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Foster-Schubert, K.E.; Overduin, J.; Prudom, C.E.; Liu, J.; Callahan, H.S.; Gaylinn, B.D.; Thorner, M.O.; Cummings, D.E. Acyl and total ghrelin are suppressed strongly by ingested proteins, weakly by lipids, and biphasically by carbohydrates. J. Clin. Endocrinol. Metab. 2008, 93, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Van Name, M.; Giannini, C.; Santoro, N.; Jastreboff, A.M.; Kubat, J.; Li, F.; Kursawe, R.; Savoye, M.; Duran, E.; Dziura, J.; et al. Blunted suppression of acyl-ghrelin in response to fructose ingestion in obese adolescents: The role of insulin resistance. Obesity 2015, 23, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Tschop, M.H.; Jarick, I.; Ehrlich, S.; Scherag, S.; Herpertz-Dahlmann, B.; Zipfel, S.; Herzog, W.; de Zwaan, M.; Burghardt, R.; et al. Genetic variation of the ghrelin activator gene ghrelin O-acyltransferase (GOAT) is associated with anorexia nervosa. J. Psychiatr. Res. 2011, 45, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Burns, S.F.; Broom, D.R.; Miyashita, M.; Mundy, C.; Stensel, D.J. A single session of treadmill running has no effect on plasma total ghrelin concentrations. J. Sports Sci. 2007, 25, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Maier, C.; Schaller, G.; Nowotny, P.; Bayerle-Eder, M.; Buranyi, B.; Luger, A.; Wolzt, M. Acute exercise has no effect on ghrelin plasma concentrations. Horm. Metab. Res. 2004, 36, 174–177. [Google Scholar] [PubMed]
- Mackelvie, K.J.; Meneilly, G.S.; Elahi, D.; Wong, A.C.; Barr, S.I.; Chanoine, J.P. Regulation of appetite in lean and obese adolescents after exercise: Role of acylated and desacyl ghrelin. J. Clin. Endocrinol. Metab. 2007, 92, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Athinarayanan, S.; Wei, R.; Zhang, M.; Bai, S.; Traber, M.G.; Yates, K.; Cummings, O.W.; Molleston, J.; Liu, W.; Chalasani, N. Genetic polymorphism of cytochrome P450 4F2, vitamin E level and histological response in adults and children with nonalcoholic fatty liver disease who participated in PIVENS and TONIC clinical trials. PLoS ONE 2014, 9, e95366. [Google Scholar] [CrossRef] [PubMed]
- Prodam, F.; Filigheddu, N. Ghrelin gene products in acute and chronic inflammation. Arch. Immunol. Ther. Exp. 2014, 62, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Delhanty, P.J.; Huisman, M.; Baldeon-Rojas, L.Y.; van den Berge, I.; Grefhorst, A.; Abribat, T.; Leenen, P.J.; Themmen, A.P.; van der Lely, A.J. Des-acyl ghrelin analogs prevent high-fat-diet-induced dysregulation of glucose homeostasis. FASEB J. 2013, 27, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Tokudome, T.; Kishimoto, I.; Miyazato, M.; Kangawa, K. Ghrelin and the cardiovascular system. Front. Horm. Res. 2014, 43, 125–133. [Google Scholar] [PubMed]
- Marchesini, G.; Pagotto, U.; Bugianesi, E.; de Iasio, R.; Manini, R.; Vanni, E.; Pasquali, R.; Melchionda, N.; Rizzetto, M. Low ghrelin concentrations in nonalcoholic fatty liver disease are related to insulin resistance. J. Clin. Endocrinol. Metab. 2003, 88, 5674–5679. [Google Scholar] [CrossRef] [PubMed]
- Mykhalchyshyn, G.; Kobyliak, N.; Bodnar, P. Diagnostic accuracy of acyl-ghrelin and it association with non-alcoholic fatty liver disease in type 2 diabetic patients. J. Diabetes Metab. Disord. 2015, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Gunta, S.S.; Mak, R.H. Ghrelin and leptin pathophysiology in chronic kidney disease. Pediatr. Nephrol. 2013, 28, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Suneja, M.; Murry, D.J.; Stokes, J.B.; Lim, V.S. Hormonal regulation of energy-protein homeostasis in hemodialysis patients: An anorexigenic profile that may predispose to adverse cardiovascular outcomes. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E55–E64. [Google Scholar] [CrossRef] [PubMed]
- Raschke, S.; Eckel, J. Adipo-myokines: Two sides of the same coin—Mediators of inflammation and mediators of exercise. Mediat. Inflamm. 2013, 2013, 320724. [Google Scholar] [CrossRef] [PubMed]
- Arias-Loste, M.T.; Ranchal, I.; Romero-Gomez, M.; Crespo, J. Irisin, a link among fatty liver disease, physical inactivity and insulin resistance. Int. J. Mol. Sci. 2014, 15, 23163–23178. [Google Scholar] [CrossRef] [PubMed]
- Anastasilakis, A.D.; Polyzos, S.A.; Saridakis, Z.G.; Kynigopoulos, G.; Skouvaklidou, E.C.; Molyvas, D.; Vasiloglou, M.F.; Apostolou, A.; Karagiozoglou-Lampoudi, T.; Siopi, A.; et al. Circulating irisin in healthy, young individuals: Day-night rhythm, effects of food intake and exercise, and associations with gender, physical activity, diet, and body composition. J. Clin. Endocrinol. Metab. 2014, 99, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.J.; Park, K.H.; Shin, S.; Zaichenko, L.; Davis, C.R.; Crowell, J.A.; Joung, H.; Mantzoros, C.S. Diet quality and diet patterns in relation to circulating cardiometabolic biomarkers. Clin. Nutr. 2016, 35, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Schlogl, M.; Piaggi, P.; Votruba, S.B.; Walter, M.; Krakoff, J.; Thearle, M.S. Increased 24-hour ad libitum food intake is associated with lower plasma irisin concentrations the following morning in adult humans. Appetite 2015, 90, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Cai, X.; Sun, Z.; Schumann, U.; Zugel, M.; Steinacker, J.M. Chronic exercise training and circulating irisin in adults: A meta-analysis. Sports Med. 2015, 45, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Zhang, X.F.; Ma, Z.M.; Pan, L.L.; Chen, Z.; Han, H.W.; Han, C.K.; Zhuang, X.J.; Lu, Y.; Li, X.J.; et al. Irisin is inversely associated with intrahepatic triglyceride contents in obese adults. J. Hepatol. 2013, 59, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Anastasilakis, A.D.; Geladari, E.V.; Mantzoros, C.S. Irisin in patients with nonalcoholic fatty liver disease. Metabolism 2014, 63, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, F.; Larson, K.; Bogardus, C.; Ravussin, E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J. Clin. Investig. 1990, 86, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Panesar, A.; Agarwal, R. Resting energy expenditure in chronic kidney disease: Relationship with glomerular filtration rate. Clin. Nephrol. 2003, 59, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Lee, S.A.; Nam, B.Y.; Park, S.; Lee, S.H.; Ryu, H.J.; Kwon, Y.E.; Kim, Y.L.; Park, K.S.; Oh, H.J.; et al. Irisin, a novel myokine is an independent predictor for sarcopenia and carotid atherosclerosis in dialysis patients. Atherosclerosis 2015, 242, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.S.; Wang, C.Y.; Lin, S.L.; Hung, K.C. Decrease in irisin in patients with chronic kidney disease. PLoS ONE 2013, 8, e64025. [Google Scholar] [CrossRef] [PubMed]
- Ebert, T.; Focke, D.; Petroff, D.; Wurst, U.; Richter, J.; Bachmann, A.; Lossner, U.; Kralisch, S.; Kratzsch, J.; Beige, J.; et al. Serum levels of the myokine irisin in relation to metabolic and renal function. Eur. J. Endocrinol. 2014, 170, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E. Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 2005, 25, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Novoselov, S.V.; Kumaraswamy, E.; Lee, B.J.; Anver, M.R.; Gladyshev, V.N.; Hatfield, D.L. Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J. Biol. Chem. 2004, 279, 8011–8017. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.E.; Zhou, J.; McMahan, W.J.; Motley, A.K.; Atkins, J.F.; Gesteland, R.F.; Burk, R.F. Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 2003, 278, 13640–13646. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L.; Schweizer, U.; Holtmann, B.; Flohe, L.; Sendtner, M.; Kohrle, J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 2003, 370, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010, 12, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Misu, H.; Ishikura, K.; Kurita, S.; Takeshita, Y.; Ota, T.; Saito, Y.; Takahashi, K.; Kaneko, S.; Takamura, T. Inverse correlation between serum levels of selenoprotein P and adiponectin in patients with type 2 diabetes. PLoS ONE 2012, 7, e34952. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.G.; Hill, K.E.; Burk, R.F. Dietary selenium intake controls rat plasma selenoprotein P concentration. J. Nutr. 1989, 119, 1010–1012. [Google Scholar] [PubMed]
- Falnoga, I.; Kobal, A.B.; Stibilj, V.; Horvat, M. Selenoprotein P in subjects exposed to mercury and other stress situations such as physical load or metal chelation treatment. Biol. Trace Elem. Res. 2002, 89, 25–33. [Google Scholar] [CrossRef]
- Huang, Z.; Rose, A.H.; Hoffmann, P.R. The role of selenium in inflammation and immunity: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2012, 16, 705–743. [Google Scholar] [CrossRef] [PubMed]
- Mattmiller, S.A.; Carlson, B.A.; Sordillo, L.M. Regulation of inflammation by selenium and selenoproteins: Impact on eicosanoid biosynthesis. J. Nutr. Sci. 2013, 2, e28. [Google Scholar] [PubMed]
- Nichol, C.; Herdman, J.; Sattar, N.; O’Dwyer, P.J.; St, J.O.R.D.; Littlejohn, D.; Fell, G. Changes in the concentrations of plasma selenium and selenoproteins after minor elective surgery: Further evidence for a negative acute phase response? Clin. Chem. 1998, 44, 1764–1766. [Google Scholar] [PubMed]
- Hesse-Bahr, K.; Dreher, I.; Kohrle, J. The influence of the cytokines Il-1beta and INFgamma on the expression of selenoproteins in the human hepatocarcinoma cell line HepG2. Biofactors 2000, 11, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Dreher, I.; Jakobs, T.C.; Kohrle, J. Cloning and characterization of the human selenoprotein P promoter. Response of selenoprotein P expression to cytokines in liver cells. J. Biol. Chem. 1997, 272, 29364–29371. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Yang, S.J.; Hwang, S.Y.; Choi, H.Y.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; Choi, K.M. Serum selenoprotein P levels in patients with type 2 diabetes and prediabetes: Implications for insulin resistance, inflammation, and atherosclerosis. J. Clin. Endocrinol. Metab. 2011, 96, E1325–E1329. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.H.; Hoffmann, P.R. Selenoproteins and cardiovascular stress. Thromb. Haemost. 2015, 113, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Hwang, S.Y.; Lee, C.H.; Hong, H.C.; Yang, S.J.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; et al. Increased selenoprotein p levels in subjects with visceral obesity and nonalcoholic Fatty liver disease. Diabetes Metab. J. 2013, 37, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; Misu, H.; Iwama, H.; Chikamoto, K.; Saito, Y.; Murao, K.; Teraguchi, A.; Lan, F.; Kikuchi, A.; Saito, R.; et al. Metformin suppresses expression of the selenoprotein P gene via an AMP-activated kinase (AMPK)/FoxO3a pathway in H4IIEC3 hepatocytes. J. Biol. Chem. 2014, 289, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E. Regulation of selenium metabolism and transport. Annu. Rev. Nutr. 2015, 35, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, W.; Dolff, S.; Benson, S.; Broecker-Preuss, M.; Behrendt, S.; Hog, A.; Fuhrer, D.; Schomburg, L.; Kohrle, J. Chronic kidney disease distinctly affects relationship between selenoprotein P status and serum thyroid hormone parameters. Thyroid 2015, 25, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.W.; Hostetter, T.H. Uremia. N. Engl. J. Med. 2007, 357, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Denecke, B.; Graber, S.; Schafer, C.; Heiss, A.; Woltje, M.; Jahnen-Dechent, W. Tissue distribution and activity testing suggest a similar but not identical function of fetuin-B and fetuin-A. Biochem. J. 2003, 376, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Mathews, S.T.; Chellam, N.; Srinivas, P.R.; Cintron, V.J.; Leon, M.A.; Goustin, A.S.; Grunberger, G. α2-HSG, a specific inhibitor of insulin receptor autophosphorylation, interacts with the insulin receptor. Mol. Cell. Endocrinol. 2000, 164, 87–98. [Google Scholar] [CrossRef]
- Stefan, N.; Hennige, A.M.; Staiger, H.; Machann, J.; Schick, F.; Krober, S.M.; Machicao, F.; Fritsche, A.; Haring, H.U. α2-Heremans-Schmid glycoprotein/fetuin-A is associated with insulin resistance and fat accumulation in the liver in humans. Diabetes Care 2006, 29, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Mathews, S.T.; Singh, G.P.; Ranalletta, M.; Cintron, V.J.; Qiang, X.; Goustin, A.S.; Jen, K.L.; Charron, M.J.; Jahnen-Dechent, W.; Grunberger, G. Improved insulin sensitivity and resistance to weight gain in mice null for the Ahsg gene. Diabetes 2002, 51, 2450–2458. [Google Scholar] [CrossRef] [PubMed]
- Siddiq, A.; Lepretre, F.; Hercberg, S.; Froguel, P.; Gibson, F. A synonymous coding polymorphism in the α2-Heremans-schmid glycoprotein gene is associated with type 2 diabetes in French Caucasians. Diabetes 2005, 54, 2477–2481. [Google Scholar] [CrossRef] [PubMed]
- Dahlman, I.; Eriksson, P.; Kaaman, M.; Jiao, H.; Lindgren, C.M.; Kere, J.; Arner, P. α2-Heremans-Schmid glycoprotein gene polymorphisms are associated with adipocyte insulin action. Diabetologia 2004, 47, 1974–1979. [Google Scholar] [CrossRef] [PubMed]
- Nimptsch, K.; Janke, J.; Pischon, T.; Linseisen, J. Association between dietary factors and plasma fetuin-A concentrations in the general population. Br. J. Nutr. 2015, 114, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Seyithanoglu, M.; Oner-Iyidogan, Y.; Dogru-Abbasoglu, S.; Tanrikulu-Kucuk, S.; Kocak, H.; Beyhan-Ozdas, S.; Kocak-Toker, N. The effect of dietary curcumin and capsaicin on hepatic fetuin-A expression and fat accumulation in rats fed on a high-fat diet. Arch. Physiol. Biochem. 2016, 122, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Malin, S.K.; Mulya, A.; Fealy, C.E.; Haus, J.M.; Pagadala, M.R.; Scelsi, A.R.; Huang, H.; Flask, C.A.; McCullough, A.J.; Kirwan, J.P. Fetuin-A is linked to improved glucose tolerance after short-term exercise training in nonalcoholic fatty liver disease. J. Appl. Physiol. 2013, 115, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Hong, H.C.; Choi, H.Y.; Yoo, H.J.; Cho, G.J.; Hwang, T.G.; Baik, S.H.; Choi, D.S.; Kim, S.M.; Choi, K.M. Effects of a three-month combined exercise programme on fibroblast growth factor 21 and fetuin-A levels and arterial stiffness in obese women. Clin. Endocrinol. 2011, 75, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Rametta, R.; Ruscica, M.; Dongiovanni, P.; Macchi, C.; Fracanzani, A.L.; Steffani, L.; Fargion, S.; Magni, P.; Valenti, L. Hepatic steatosis and PNPLA3 I148M variant are associated with serum Fetuin-A independently of insulin resistance. Eur. J. Clin. Investig. 2014, 44, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Yonal, O.; Kurt, R.; Ari, F.; Oral, A.Y.; Celikel, C.A.; Korkmaz, S.; Ulukaya, E.; Ozdogan, O.; Imeryuz, N.; et al. Serum fetuin A/α2HS-glycoprotein levels in patients with non-alcoholic fatty liver disease: Relation with liver fibrosis. Ann. Clin. Biochem. 2010, 47, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.Y.; Yang, Y.C.; Wu, H.T.; Wu, J.S.; Lu, F.H.; Chang, C.J. Increased fetuin-A concentrations in impaired glucose tolerance with or without nonalcoholic fatty liver disease, but not impaired fasting glucose. J. Clin. Endocrinol. Metab. 2012, 97, 4717–4723. [Google Scholar] [CrossRef] [PubMed]
- Cottone, S.; Palermo, A.; Arsena, R.; Riccobene, R.; Guarneri, M.; Mule, G.; Tornese, F.; Altieri, C.; Vaccaro, F.; Previti, A.; et al. Relationship of fetuin-A with glomerular filtration rate and endothelial dysfunction in moderate-severe chronic kidney disease. J. Nephrol. 2010, 23, 62–69. [Google Scholar] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petta, S.; Gastaldelli, A.; Rebelos, E.; Bugianesi, E.; Messa, P.; Miele, L.; Svegliati-Baroni, G.; Valenti, L.; Bonino, F. Pathophysiology of Non Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 2082. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122082
Petta S, Gastaldelli A, Rebelos E, Bugianesi E, Messa P, Miele L, Svegliati-Baroni G, Valenti L, Bonino F. Pathophysiology of Non Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2016; 17(12):2082. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122082
Chicago/Turabian StylePetta, Salvatore, Amalia Gastaldelli, Eleni Rebelos, Elisabetta Bugianesi, Piergiorgio Messa, Luca Miele, Gianluca Svegliati-Baroni, Luca Valenti, and Ferruccio Bonino. 2016. "Pathophysiology of Non Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 17, no. 12: 2082. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122082