
Rapid Identification of Pathogenic Variants in Two Cases of Charcot-Marie-Tooth Disease by Gene-Panel Sequencing

Abstract
:1. Introduction
2. Results
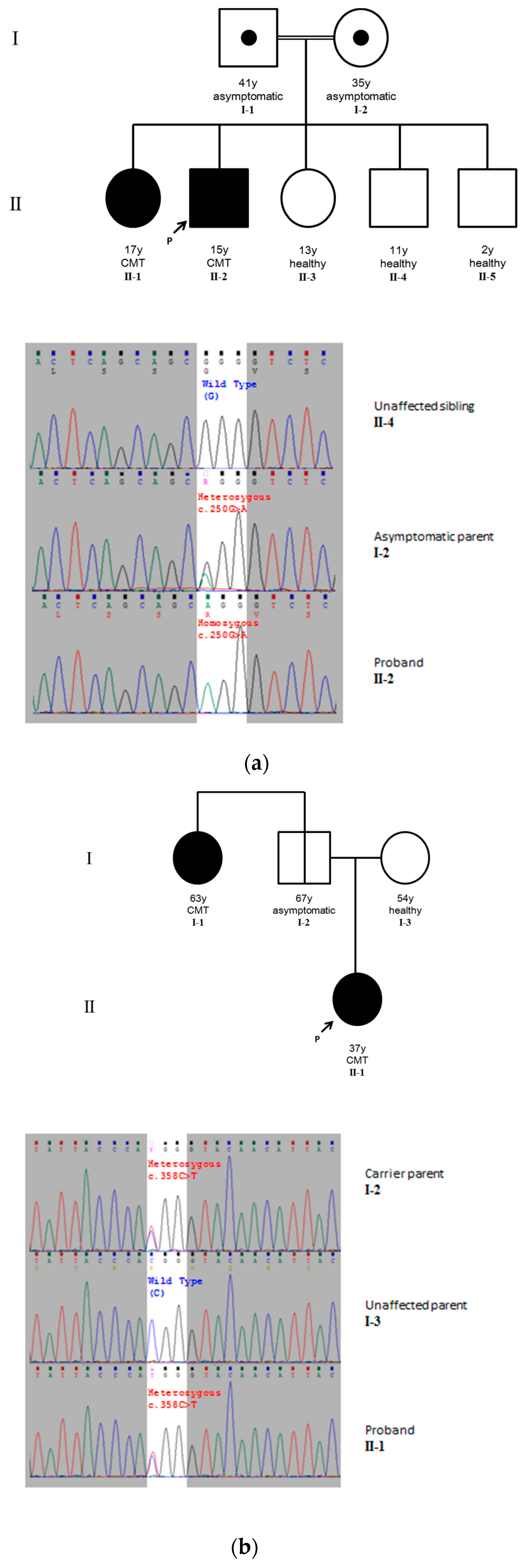
2.1. Case Report
2.1.1. Case 1
2.1.2. Case 2
2.2. Identification of Pathogenic Variants
2.2.1. Case 1
2.2.2. Case 2
2.3. Estimation of Minimal Sequencing Depth for Genetic Diagnosis
3. Discussion
4. Materials and Methods
4.1. Patients and Human Ethics
4.2. Gene-Panel and Next-Generation Sequencing
4.3. Variant Discovery and Annotation
4.4. Pathogenic Variant Validation and Cascade Screening
4.5. Down-Sampling Experiment
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CMT | Charcot-Marie-Tooth Disease |
| DI-CMT | Dominant intermediate CMT |
References
- Braathen, G.J. Genetic epidemiology of Charcot-Marie-Tooth disease. Acta Neurol. Scand. Suppl. 2012, 126, iv-22. [Google Scholar] [CrossRef] [PubMed]
- Rossor, A.M.; Polke, J.M.; Houlden, H.; Reilly, M.M. Clinical implications of genetic advances in Charcot–Marie–Tooth disease. Nat. Rev. Neurol. 2013, 9, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.P.; Emery, A.E. A family study of Charcot-Marie-Tooth disease. J. Med. Genet. 1982, 19, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, L.R.; Zwi, M.B.; McLeod, J.G.; Nicholson, G.A. Chromosome I linkage studies in Charcot-Marie-Tooth neuropathy type I. Am. J. Hum. Genet. 1988, 42, 756–771. [Google Scholar] [PubMed]
- Gabreëls-Festen, A.A.; Gabreëls, F.J.; Jennekens, F.G.; Joosten, E.M.; Janssen-van Kempen, T.W. Autosomal recessive form of hereditary motor and sensory neuropathy type I. Neurology 1992, 42, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Cornell, J.; Sellars, S.; Beighton, P. Autosomal recessive inheritance of Charcot-Marie-Tooth disease associated with sensorineural deafness. Clin. Genet. 1984, 25, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Ionasescu, V.V.; Burns, T.L.; Searby, C.; Ionasescu, R. X-linked dominant Charcot-Marie-Tooth neuropathy with 15 cases in a family genetic linkage study. Muscle Nerve 1988, 11, 1154–1156. [Google Scholar] [CrossRef] [PubMed]
- Ionasescu, V.V.; Trofatter, J.; Haines, J.L.; Summers, A.M.; Ionasescu, R.; Searby, C. Heterogeneity in X-linked recessive Charcot-Marie-Tooth neuropathy. Am. J. Hum. Genet. 1991, 48, 1075–1083. [Google Scholar] [PubMed]
- Meggouh, F.; Bienfait, H.M.E.; Weterman, M.A.J.; de Visser, M.; Baas, F. Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology 2006, 67, 1476–1478. [Google Scholar] [CrossRef] [PubMed]
- James, P.; Rankin, J.; Talbot, K. Asymmetrical late onset motor neuropathy associated with a novel mutation in the small heat shock protein HSPB1 (HSP27). J. Neurol. Neurosurg. Psychiatry 2008, 79, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Pla-Martin, D.; Calpena, E.; Lupo, V.; Marquez, C.; Rivas, E.; Sivera, R.; Sevilla, T.; Palau, F.; Espinos, C. Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum. Mol. Genet. 2015, 24, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Østern, R.; Fagerheim, T.; Hjellnes, H.; Nygård, B.; Mellgren, S.I.; Nilssen, Ø. Diagnostic laboratory testing for Charcot Marie Tooth disease (CMT): The spectrum of gene defects in Norwegian patients with CMT and its implications for future genetic test strategies. BMC Med. Genet. 2013, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schöneborn, S.; Tölle, D.; Senderek, J.; Eggermann, K.; Elbracht, M.; Kornak, U.; von der Hagen, M.; Kirschner, J.; Leube, B.; Müller-Felber, W.; et al. Diagnostic algorithms in Charcot-Marie-Tooth neuropathies: Experiences from a German genetic laboratory on the basis of 1206 index patients. Clin. Genet. 2016, 89, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.M.; Choi, J.H.; Lee, S.Y.; Yoo, N.C. Applications of DNA microarray in disease diagnostics. J. Microbiol. Biotechnol. 2009, 19, 635–646. [Google Scholar] [PubMed]
- Baaj, Y.; Magdelaine, C.; Ubertelli, V.; Valat, C.; Mousseau, Y.; Qiu, H.; Funalot, B.; Vallat, J.-M.; Sturtz, F.G. Multiplex Detection and Genotyping of Point Mutations Involved in Charcot-Marie-Tooth Disease Using a Hairpin Microarray-Based Assay. Res. Lett. Biochem. 2009, 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, J.C.; Isfort, M.C.; Roggenbuck, J.; Arnold, W.D. The genetics of Charcot-Marie-Tooth disease: Current trends and future implications for diagnosis and management. Appl. Clin. Genet. 2015, 8, 235–243. [Google Scholar] [PubMed]
- Gonzaga-Jauregui, C.; Harel, T.; Gambin, T.; Kousi, M.; Griffin, L.B.; Francescatto, L.; Ozes, B.; Karaca, E.; Jhangiani, S.N.; Bainbridge, M.N.; et al. Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep. 2015, 12, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, V.V.; Sudnitsyna, M.V.; Strelkov, S.V.; Gusev, N.B. Structure and properties of G84R and L99M mutants of human small heat shock protein HspB1 correlating with motor neuropathy. Arch. Biochem. Biophys. 2013, 538, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Ammar, N.; Nelis, E.; Merlini, L.; Barisić, N.; Amouri, R.; Ceuterick, C.; Martin, J.J.; Timmerman, V.; Hentati, F.; De Jonghe, P. Identification of novel GDAP1 mutations causing autosomal recessive Charcot-Marie-Tooth disease. Neuromuscul. Disord. NMD 2003, 13, 720–728. [Google Scholar] [CrossRef]
- Claramunt, R.; Pedrola, L.; Sevilla, T.; López de Munain, A.; Berciano, J.; Cuesta, A.; Sánchez-Navarro, B.; Millán, J.M.; Saifi, G.M.; Lupski, J.R.; et al. Genetics of Charcot-Marie-Tooth disease type 4A: Mutations, inheritance, phenotypic variability, and founder effect. J. Med. Genet. 2005, 42, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Zimon, M.; Baets, J.; Fabrizi, G.M.; Jaakkola, E.; Kabzinska, D.; Pilch, J.; Schindler, A.B.; Cornblath, D.R.; Fischbeck, K.H.; Auer-Grumbach, M.; et al. Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 2011, 77, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Laššuthová, P.; Šafka Brožková, D.; Krůtová, M.; Neupauerová, J.; Haberlová, J.; Mazanec, R.; Dřímal, P.; Seeman, P. Improving diagnosis of inherited peripheral neuropathies through gene panel analysis. Orphanet J. Rare Dis. 2016, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; Magri, S.; Bagarotti, A.; Carecchio, M.; Piscosquito, G.; Pareyson, D.; Varrasi, C.; Vecchio, D.; Zonta, A.; Cantello, R.; et al. A novel synonymous mutation in the MPZ gene causing an aberrant splicing pattern and Charcot-Marie-Tooth disease type 1b. Neuromuscul. Disord. 2016, 26, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Lupo, V.; García-García, F.; Sancho, P.; Tello, C.; García-Romero, M.; Villarreal, L.; Alberti, A.; Sivera, R.; Dopazo, J.; Pascual-Pascual, S.I.; et al. Assessment of Targeted Next-Generation Sequencing as a Tool for the Diagnosis of Charcot-Marie-Tooth Disease and Hereditary Motor Neuropathy. J. Mol. Diagn. 2016, 18, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.H.; Hong, Y.B.; Hyun, Y.S.; Nam, D.E.; Kwak, G.; Hwang, S.H.; Choi, B.-O.; Chung, K.W. Identification of Genetic Causes of Inherited Peripheral Neuropathies by Targeted Gene Panel Sequencing. Mol. Cells 2016, 39, 382–388. [Google Scholar] [PubMed]
- Miller, L.J.; Saporta, A.S.D.; Sottile, S.L.; Siskind, C.E.; Feely, S.M.E.; Shy, M.E. Strategy for genetic testing in Charcot-Marie-disease. Acta Myol. 2011, 30, 109–116. [Google Scholar] [PubMed]
- Banchs, I.; Casasnovas, C.; Albertí, A.; De Jorge, L.; Povedano, M.; Montero, J.; Martínez-Matos, J.A.; Volpini, V. Diagnosis of Charcot-Marie-Tooth disease. J. Biomed. Biotechnol. 2009, 2009, 985415. [Google Scholar] [CrossRef] [PubMed]
- De Jonghe, P.; Nelis, E.; Timmerman, V.; Löfgren, A.; Martin, J.J.; Van Broeckhoven, C. Molecular diagnostic testing in Charcot-Marie-Tooth disease and related disorders. Approaches and results. Ann. N. Y. Acad. Sci. 1999, 883, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.; Sebire, N.; Harris, J.; Robinson, S.; Regan, L. The risks associated with pregnancy in women aged 35 years or older. Hum. Reprod. Oxf. Engl. 2000, 15, 2433–2437. [Google Scholar] [CrossRef]
- Gray, E.; Eden, M.; Vass, C.; McAllister, M.; Louviere, J.; Payne, K. Valuing Preferences for the Process and Outcomes of Clinical Genetics Services: A Pilot Study. Patient 2016, 9, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Musters, A.M.; Twisk, M.; Leschot, N.J.; Oosterwijk, C.; Korevaar, J.C.; Repping, S.; van der Veen, F.; Goddijn, M. Perspectives of couples with high risk of transmitting genetic disorders. Fertil. Steril. 2010, 94, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Carson, A.R.; Smith, E.N.; Matsui, H.; Brækkan, S.K.; Jepsen, K.; Hansen, J.-B.; Frazer, K.A. Effective filtering strategies to improve data quality from population-based whole exome sequencing studies. BMC Bioinform. 2014, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Field, M.A.; Cho, V.; Andrews, T.D.; Goodnow, C.C. Reliably Detecting Clinically Important Variants Requires Both Combined Variant Calls and Optimized Filtering Strategies. PLoS ONE 2015, 10, e0143199. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.-B.; Lee, I.-H.; Park, J.-H.; Hambuch, T.; Choe, Y.; Kim, M.; Lee, K.; Song, T.; Neu, M.B.; Gupta, N.; et al. Reducing false-positive incidental findings with ensemble genotyping and logistic regression based variant filtering methods. Hum. Mutat. 2014, 35, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef] [PubMed]
- Cacheiro, P.; Ordóñez-Ugalde, A.; Quintáns, B.; Piñeiro-Hermida, S.; Amigo, J.; García-Murias, M.; Pascual-Pascual, S.I.; Grandas, F.; Arpa, J.; Carracedo, A.; et al. Evaluating the Calling Performance of a Rare Disease NGS Panel for Single Nucleotide and Copy Number Variants. Mol. Diagn. Ther. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.E.; Thomas, P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain J. Neurol. 1980, 103, 259–280. [Google Scholar] [CrossRef]
- Pareyson, D.; Scaioli, V.; Laurà, M. Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease. Neuromol. Med. 2006, 8, 3–22. [Google Scholar] [CrossRef]
- Nicholson, G.; Myers, S. Intermediate forms of Charcot-Marie-Tooth neuropathy: A review. Neuromolecul. Med. 2006, 8, 123–130. [Google Scholar] [CrossRef]
- Cornett, K.M.D.; Menezes, M.P.; Bray, P.; Halaki, M.; Shy, R.R.; Yum, S.W.; Estilow, T.; Moroni, I.; Foscan, M.; Pagliano, E.; et al. Inherited Neuropathies Consortium Phenotypic Variability of Childhood Charcot-Marie-Tooth Disease. JAMA Neurol. 2016, 73, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Saporta, A.S.D.; Sottile, S.L.; Miller, L.J.; Feely, S.M.E.; Siskind, C.E.; Shy, M.E. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 2011, 69, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv. 2013. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Wang, K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Gui, H.S.; Kwan, J.S.; Bao, S.Y.; Sham, P.C. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 2012, 40, gkr1257. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Poon, W.T.; Chan, K.Y.; Au, K.M.; Tong, S.F.; Chan, Y.W.; Lam, C.W.; Chow, C.B. Novel missense mutation (Y279S) in the GLRA1 gene causing hyperekplexia. Clin. Chim. Acta 2006, 364, 361–362. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | CMT Phenotype | Inheritance | Chromosome Location |
|---|---|---|---|
| KIF1B | CMT 2A1 | AD | 1p36.22 |
| MFN2 | CMT 2A2A & 2A2B | AD, AR | 1p36.22 |
| YARS | Dominant intermediate CMT (DI-CMT) type C | AD | 1p35.1 |
| LMNA | CMT 2B1 | AR | 1q22 |
| MPZ | DI-CMT type D, CMT 1B, 2I & 2J | AD | 1q23.3 |
| RAB7A | CMT 2B | AD | 3q21.3 |
| SH3TC2 | CMT 4C | AR | 5q32 |
| FIG4 | CMT 4J | AR | 6q21 |
| GARS | CMT 2D | AD | 7p14.3 |
| HSPB1 | CMT 2F | AD | 7q11.23 |
| NEFL | CMT 1F & 2E | AD, AR | 8p21.2 |
| GDAP1 | Recessive intermediate CMT type A, CMT 2K & 4A | AD, AR | 8p21.11 |
| NDRG1 | CMT 4D | AR | 8q24.22 |
| EGR2 | CMT 1D | AD | 10q21.3 |
| SBF2 | CMT 4B2 | AR | 11p15.4 |
| MTMR2 | CMT 4B1 | AR | 11q21 |
| FGD4 | CMT 4H | AR | 12p11.21 |
| TRPV4 | Hereditary motor and sensory neuropathy (HMSN) IIc | AD | 12q24.11 |
| HSPB8 | CMT 2L | AD | 12q24.23 |
| LITAF | CMT 1C | AD | 16p13.13 |
| AARS | CMT 2N | AD | 16q22.1 |
| PMP22 | CMT 1A & 1E | AD | 17p12 |
| DNM2 | DI-CMT type B, CMT 2M | AD | 19p13.2 |
| PRX | CMT 4F | AR | 19q13.2 |
| MED25 | CMT 2B2 | AR | 19q13.33 |
| GJB1 | X-linked dominant CMT type 1 | XLD | Xq13.1 |
| PRPS1 | X-lined recessive CMT type 5 | XLR | Xq22.3 |
| Case | Variant | Gene (Variant Type) | wANNOVAR (Exome Aggregation Consortium (ExAC) Overall Minor Allele Frequency (MAF)) | MutationTaster Prediction (Prediction Probability, Pcorrect) | KGGSeq Prediction (Disease-Casual Probability, Pdisease) |
|---|---|---|---|---|---|
| 1 | chr1:156109095_156109095delA LMNA:NM_170707:cDNA.2405_2405delA | Lamin A/C (Heterozygous 3′ UTR indel in a poly-A stretch) * | (excluded) 1 | Disease-causing (Pcorrect > 0.999) | No prediction 2 |
| chr7:75932279G>A HSPB1:NM_001540:c.G250A (p.G84R) | Heat shock protein family B member 1 (Homozygous nonsynonymous SNP) | Shortlisted (no ExAC data) | Disease-causing (Pcorrect > 0.999) | Disease-causing (Pdisease = 0.681) | |
| chr11:9861208G>C SBF2:NM_030962:c.C3292G (p.L1098V) | SET binding factor 2 (Heterozygous nonsynonymous SNP) | Shortlisted (MAF = 0.0209) | Polymorphism (Pcorrect = 0.054) 3 | Non-disease-causing (Pdisease = 3.46 × 10−4) | |
| chr11:95595177A>G MTMR2:NM_016156: c.T447C (p.Y149Y) | Myotubularin related protein 2 (Heterozygous synonymous SNP) | Shortlisted (MAF = 2.527 × 10−5, all from South Asian data in ExAC) | Disease-causing (Pcorrect = 1) | No prediction 2 | |
| 2 | chr1:10342522G>A KIF1B:NM_015074:c.G1227A (p.T409T) | Kinesin family member 1B (Heterozygous synonymous SNP) | Shortlisted (MAF = 0.0328) | Polymorphism (Pcorrect = 2.98 × 10−17) 3 | (filtered) 4 |
| chr1:10397567A>G KIF1B:NM_015074:c.A3260G (p.Y1087C) | Kinesin family member 1B (Heterozygous synonymous SNP) | Shortlisted (MAF = 0.0325) | Polymorphism (Pcorrect = 5.41 × 10−11) 3 | Non-disease-causing (Pdisease = 0.039) | |
| chr1:156109095_156109095delA LMNA: NM_170707:cDNA.2405_2405delA | Lamin A/C (Heterozygous 3′ UTR indel in a poly-A stretch) * | (excluded) 1 | Disease-causing (Pcorrect > 0.999) | No prediction 2 | |
| chr8:75272419C>T GDAP1:NM_018972:c.C358T (p.R120W) | Ganglioside-induced differentiation-associated protein 1 (Heterozygous nonsynonymous SNP) | Shortlisted (no ExAC data) | Disease-causing (Pcorrect > 0.999) | Disease-causing (Pdisease = 0.500) | |
| chr11:9990017G>A SBF2:NM_030962:c.C1471T (p.L491F) | SET binding factor 2 (Heterozygous nonsynonymous SNP) | Shortlisted (no ExAC data) | Disease-causing (Pcorrect > 0.999) | Non-disease-causing (Pdisease = 0.044) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, C.-C.; Tai, S.-M.; Lee, E.C.-N.; Mak, T.S.-H.; Liu, T.K.-T.; Tang, V.W.-L.; Poon, W.-T. Rapid Identification of Pathogenic Variants in Two Cases of Charcot-Marie-Tooth Disease by Gene-Panel Sequencing. Int. J. Mol. Sci. 2017, 18, 770. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18040770
Ho C-C, Tai S-M, Lee EC-N, Mak TS-H, Liu TK-T, Tang VW-L, Poon W-T. Rapid Identification of Pathogenic Variants in Two Cases of Charcot-Marie-Tooth Disease by Gene-Panel Sequencing. International Journal of Molecular Sciences. 2017; 18(4):770. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18040770
Chicago/Turabian StyleHo, Chi-Chun, Shuk-Mui Tai, Edmond Chi-Nam Lee, Timothy Shin-Heng Mak, Timothy Kam-Tim Liu, Victor Wai-Lun Tang, and Wing-Tat Poon. 2017. "Rapid Identification of Pathogenic Variants in Two Cases of Charcot-Marie-Tooth Disease by Gene-Panel Sequencing" International Journal of Molecular Sciences 18, no. 4: 770. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18040770