2.1. Performance of the Hotspot Prediction
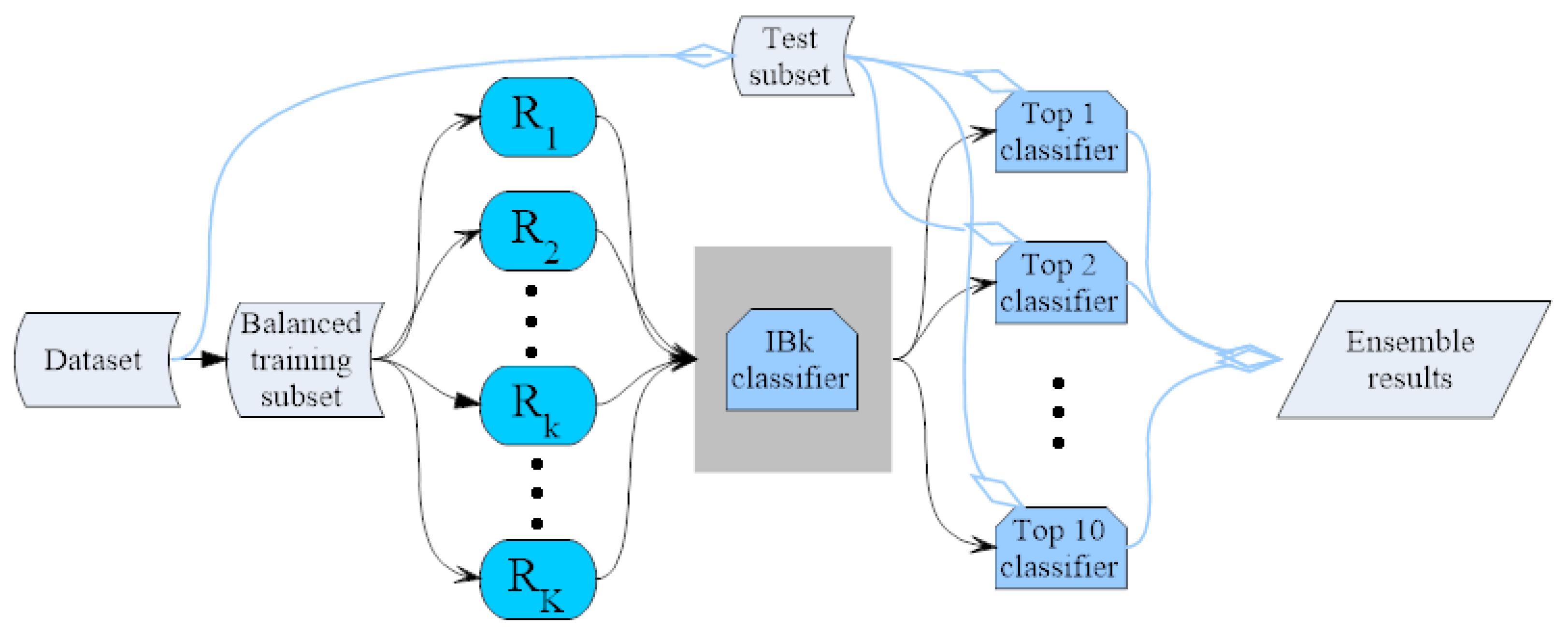
In the running of the random projection-based classifier, different random projections in Equation (1) construct different classifiers. After running the classifier 100 times, 100 classifiers with random projections R are formed and trained on the training subset . As a result, 100 predictions are obtained. All of the classifiers are ranked in terms of the prediction measure F1. The ensemble of several top N classifiers is then tested on the test subset . In this work, the ASEdb0 is regarded as the training dataset, and the test dataset is BID0; while the predictions on the ASEdb0 dataset are also tested by training on the BID0 dataset.
Table 1 shows the performance of the top individual classifiers trained by the ASEdb0 dataset and the prediction performance on the BID0 dataset. The individual classifiers are ranked in terms of the
F1 measure in the training process. The top classifiers yield good predictions on the BID0 dataset. It achieves an F1 of 0.109, as well as a precision of 0.069 at a sensitivity of 0.259 in the training process and, therefore, yields an F1 of 0.315, as well as a precision of 0.220 at a sensitivity of 0.558 in the test process. Here, the dimensionality of the original data is reduced from 7072 to only five.
Table 2 shows the performance comparison of the ensembles of the top
N classifiers. In the classifier ensemble, the majority vote technique was applied to the ensemble, i.e., one residue will be identified as the hotspot if half of the
N classifiers predict it to be the hotspot. Here, seven ensembles of the number of top classifiers are listed, i.e., 2, 3, 5, 10, 15, 25 and 50. From
Table 2, it can be seen that the ensemble of the top three classifiers with the majority vote yields a good performance compared with other classifier ensembles. It yields an
MCC (Matthews Correlation Coefficient) of 0.428, as well as a precision of 0.245 at a sensitivity of 0.793, for testing on the ASEdb0 dataset by training on the BID0 dataset; and it yields an
MCC of 0.601, as well as a precision of 0.440 at a sensitivity of 0.846, for testing on the BID0 dataset by training on the ASEdb0 dataset. The ensemble of the top three classifiers resulted in a dramatic improvement, compared with the top three individual classifiers. The reason for the improvement is most likely in that a suitable random projection makes the classifier more diverse, where the detailed results are not shown here. Previous methods also showed that the ensemble of more diverse classifiers yielded more efficient predictions [
30].
It seems that the more top classifiers the ensemble contains, the worse the ensemble performs. The ensemble with the top 50 classifiers performs the worst both for testing on the ASEdb0 and the BID0 dataset. Therefore, a suitable number of top classifiers can improve the predictions of hotspot residues. Moreover, our method on the BID0 dataset performs better than that on the ASEdb0 dataset, maybe because of the larger ratio of hotspots to the total residues in BID0 (1.831%) than that in ASEdb0 (1.445%).
Furthermore, the performance comparison of ensembles with different numbers of reduced instance dimensions by the random projection technique was investigated. The ensembles of random projections with seven reduced dimensions were built, i.e., the dimensions of 1, 2, 5, 10, 20, 50 and 100. The ensemble with the reduced dimension of five performs the best among the seven ensembles, while the ensemble of the top three classifiers with instance dimension of one also performs well in the hotspot predictions for the whole sequences of proteins, which yields an
MCC of 0.475, as well as a precision of 0.704 at a sensitivity of 0.328.
Table 3 shows the performance comparison of the classifier ensemble with different numbers of reduced dimensions on the BID0 test dataset.
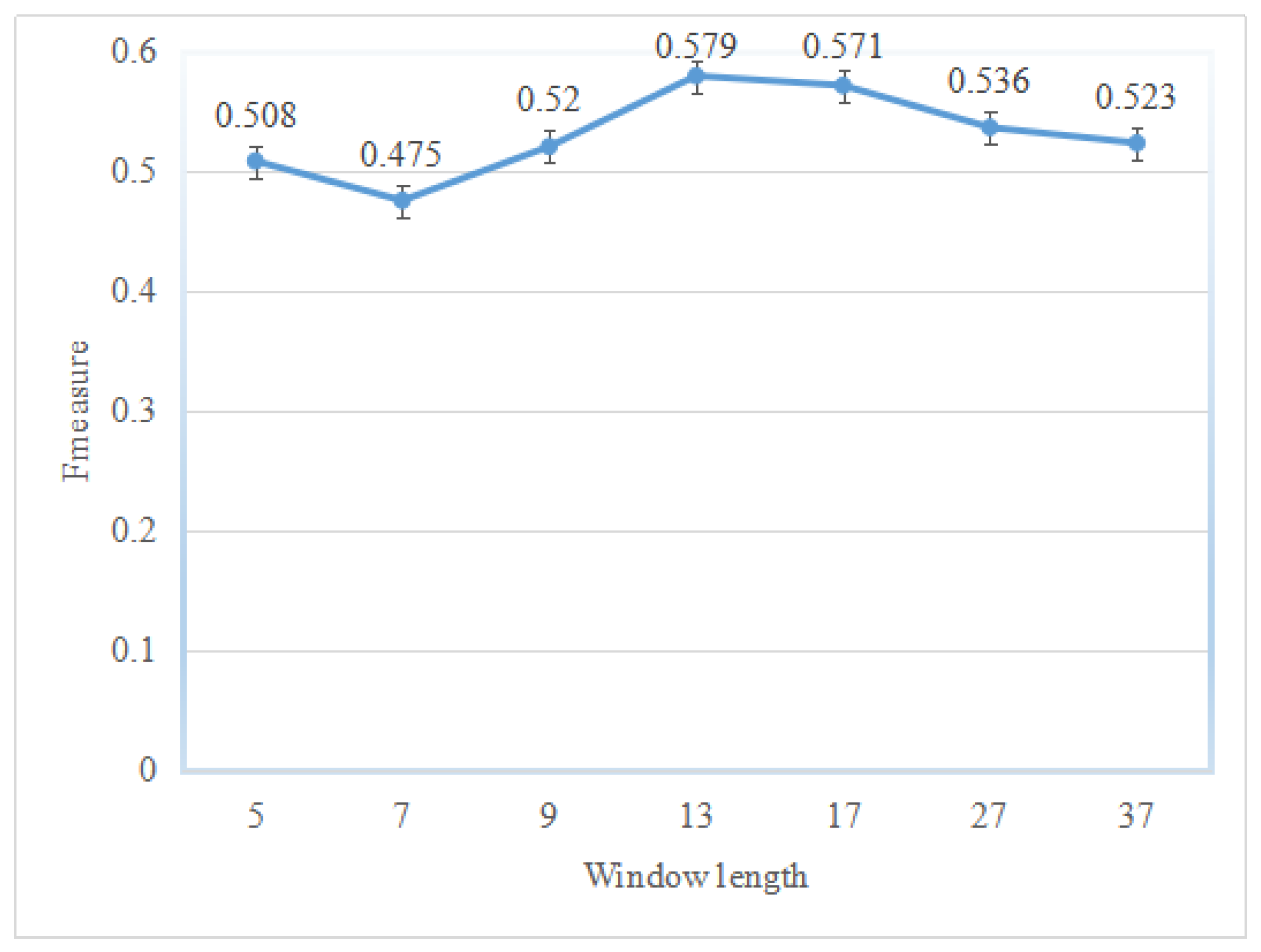
This study adopted the window length technique to encode input instances of classifiers; however, the sliding window technique makes the performance of the classifier varied. To show which window length makes the classifiers better for a specific type of dataset, several windows with different lengths were investigated.
Figure 1 shows the prediction performance on different sliding windows on the BID0 dataset. Among the seven sliding windows, the window with length 13 performs the best, which yields an
F1 of 0.579. It should be mentioned here that classifier ensembles with a suitable window length perform better than those with a smaller or bigger length.
2.2. Comparison with Other Methods
So far, few works identified hotspots from the whole protein sequences by sequence information alone. Some top hotspot predictors did the predictions based on protein structures. Most of hotspot prediction methods predicted hotspots from protein-protein interfaces or from some benchmark datasets, such as ASEdb0 and BID0, which contained approximately the same hotspots and non-hotspots. Therefore, the random predictor is used to compare with our method. The random predictor was run 100 times, and the average performance was calculated. Furthermore, for prediction comparison, the tool of ISIS (Interaction Sites Identified from Sequence) [
20] on the PredictProtein server [
31] was adopted, which has been applied in hotspot predictions on the dataset of interface residues [
20]. ISIS is a machine learning-based method that identified interacting residues from the sequence alone. Similar to our method, although the method was developed using transient protein-protein interfaces from complexes of experimentally-known 3D structures, it only used the sequence and predicted 2D information. In PredictProtein, it predicted a residue as a hotspot if the prediction score of the residue was bigger than 21, otherwise being non-hotspot residues. Since PredictProtein currently cannot process short input sequences less than 17 residues, protein sequences in PDB names “1DDMB” and “2NMBB” were removed from the BID0 test set. We tested all of the sequences of more than 17 residues on the BID0 dataset, and the performance of hotspot predictions on the dataset was obtained. The predictions of ISIS method can be referred to the
Supplementary Materials.
Table 4 lists the hotspot prediction comparison in detail. Our method developed a random projection ensemble system yielding a final precision of 0.440 at a sensitivity of 0.846 by the use of sequence information only. Results showed that our method outperforms the random predictor. Furthermore, our method outperformed the ISIS method. Actually, ISIS was developed to identify protein-protein interactions. The power of ISIS for the identification of hotspot residues was poor. It can predict nine of 47 real hotspots correctly; however, 2920 non-hotspots were predicted to be hotspots in the BID0 dataset.
We also show the performance of classifier ensemble in several measures based on the measure of sensitivity.
Figure 2 illustrates the performance of the ensemble classifier with the majority vote for the test set BID0. Although it is very difficult to identify hotspots from the whole protein sequences, our method yields a good result based on sequence information only.
2.3. Case Study of Hotspot Predictions
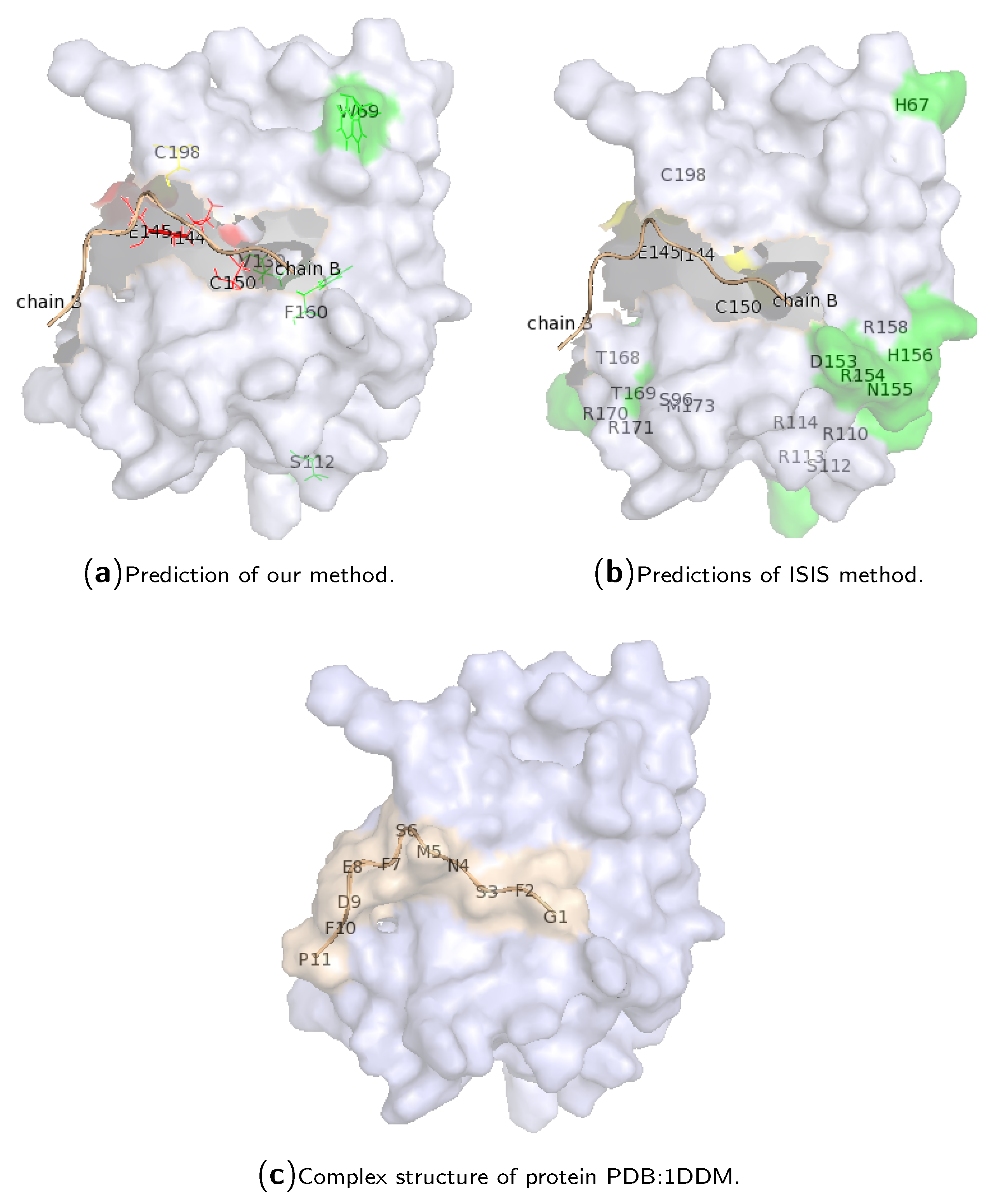
To show the performance of our method on a single protein chain, hotspot predictions for chain “A” of protein PDB:1DDM are illustrated in
Figure 3. Protein 1DDM is an in vivo complex containing a phosphotyrosine-binding (PTB) domain (chain “A”) of the cell fate determinant Numb, which can bind a diverse array of peptide sequences in vitro, and a peptide containing an amino acid sequence “NMSF” derived from the Numb-associated kinase (Nak) (chain “B”). The Numb PTB domain is in complex with the Nak peptide. The chain “A” contains 135 amino acid residues, where only residues E144, I145, C150 and C198 are real hotspot residues in complex with the chain “B” of the protein (which contains 11 amino acid residues; see
Figure 3c). Our method correctly predicted the first three true hotspots, and hotspot residue 198 was predicted as a non-hotspot, while residues 69, 112, 130 and 160 were wrongly predicted as hotspot residues. All of them are located at the surface of the protein structure. The results of ISIS are also illustrated in
Figure 3b. The ISIS method cannot identify the four true hotspot residues, although most of the hotspot predictions are located at the surface of the protein.







{kind=link}
{kind=link}
{kind=link}
{kind=link}