Telocinobufagin, a Novel Cardiotonic Steroid, Promotes Renal Fibrosis via Na+/K+-ATPase Profibrotic Signaling Pathways

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. In Vivo Studies
2.2. Assessment of Blood Pressure and Renal Function
2.3. Reagents, Cell Culture, and Quantitative Polymerase Chain Reaction (PCR)
2.4. Quantitative Histology
2.5. Patient Study
2.6. Biochemical Assays
2.7. Statistical Analysis
3. Results
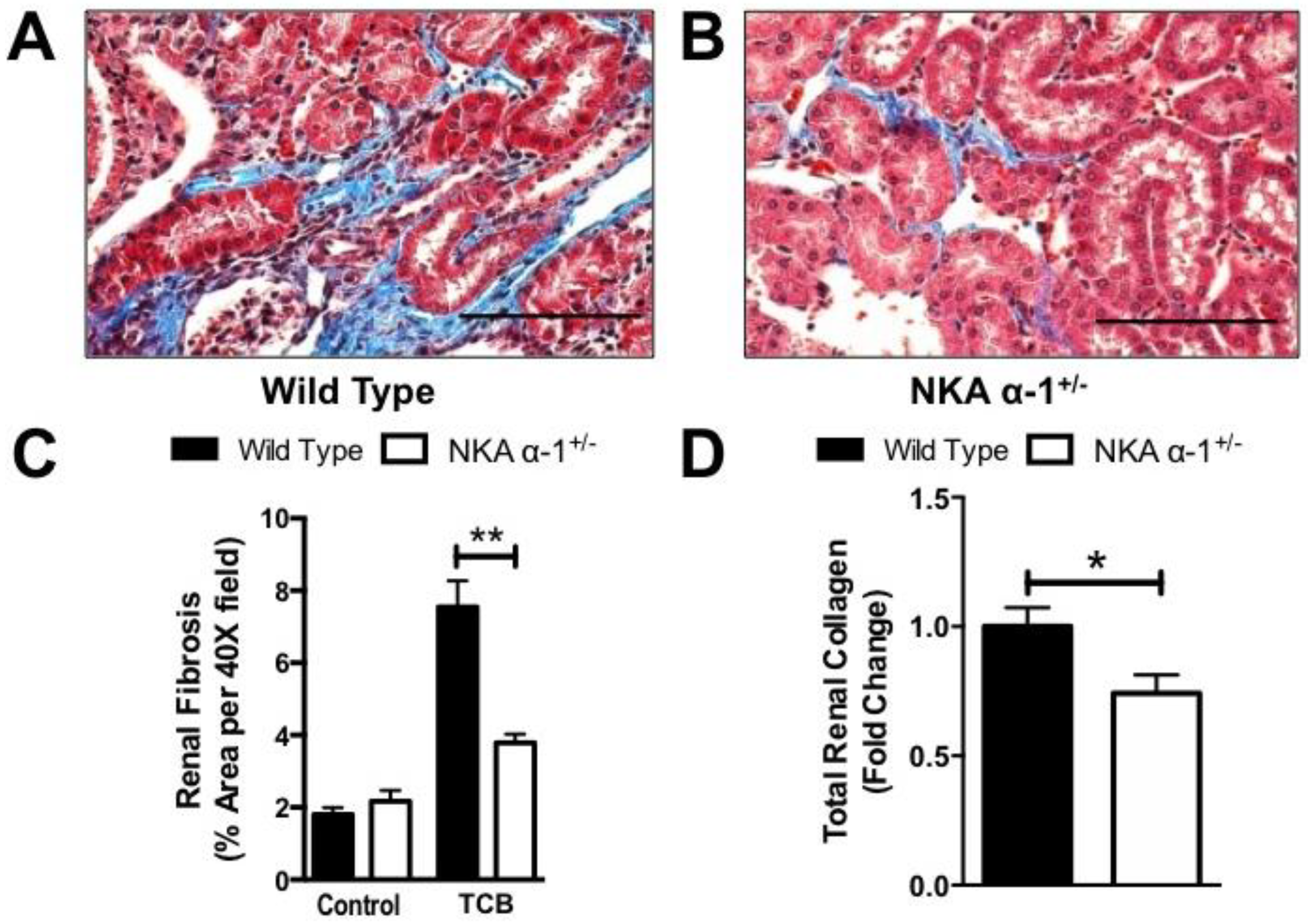
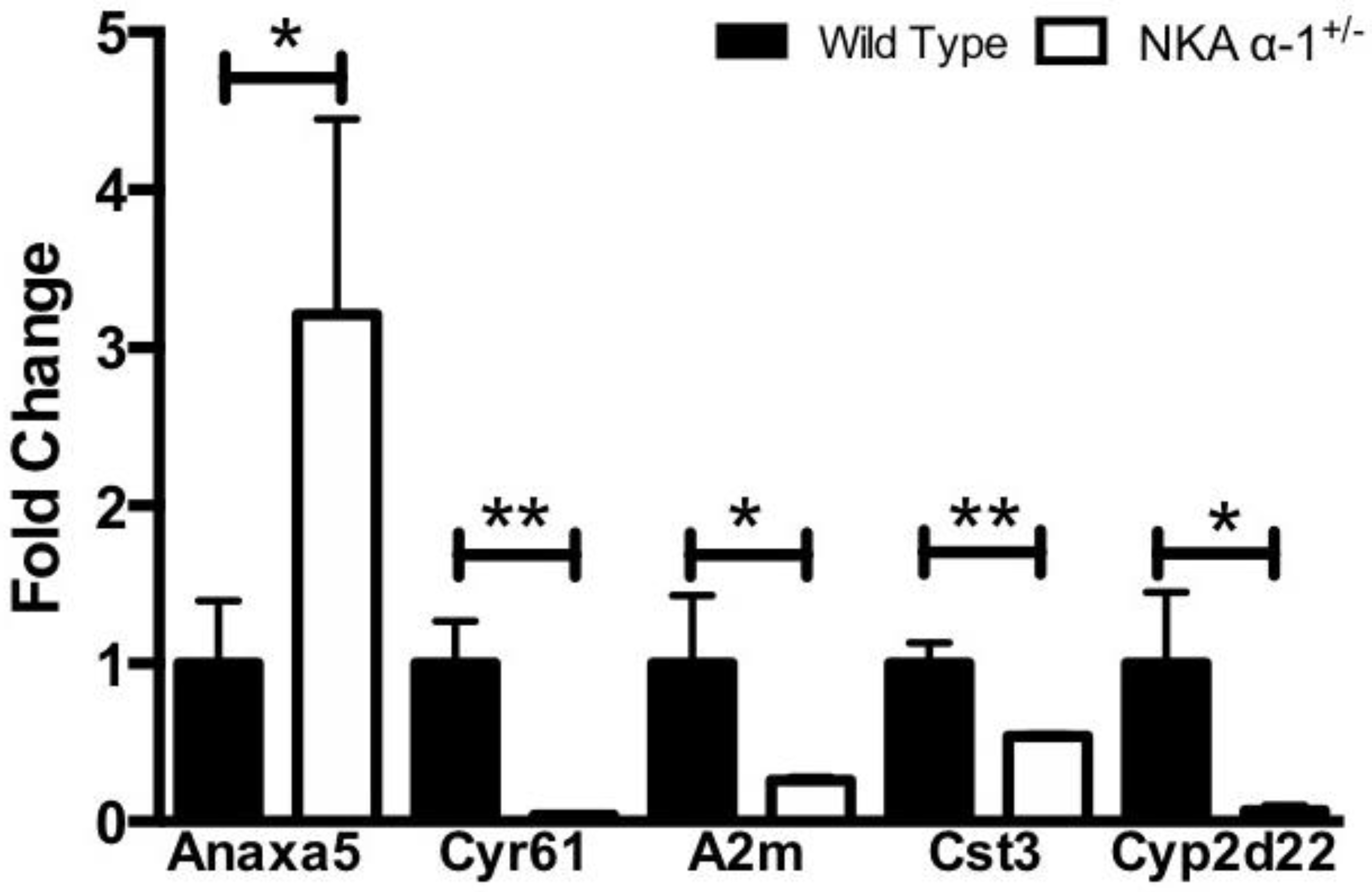
3.1. Telocinobufagin-Induced Renal Fibrosis and Dysfunction Depends on Na+/K+-ATPase Signaling In Vivo
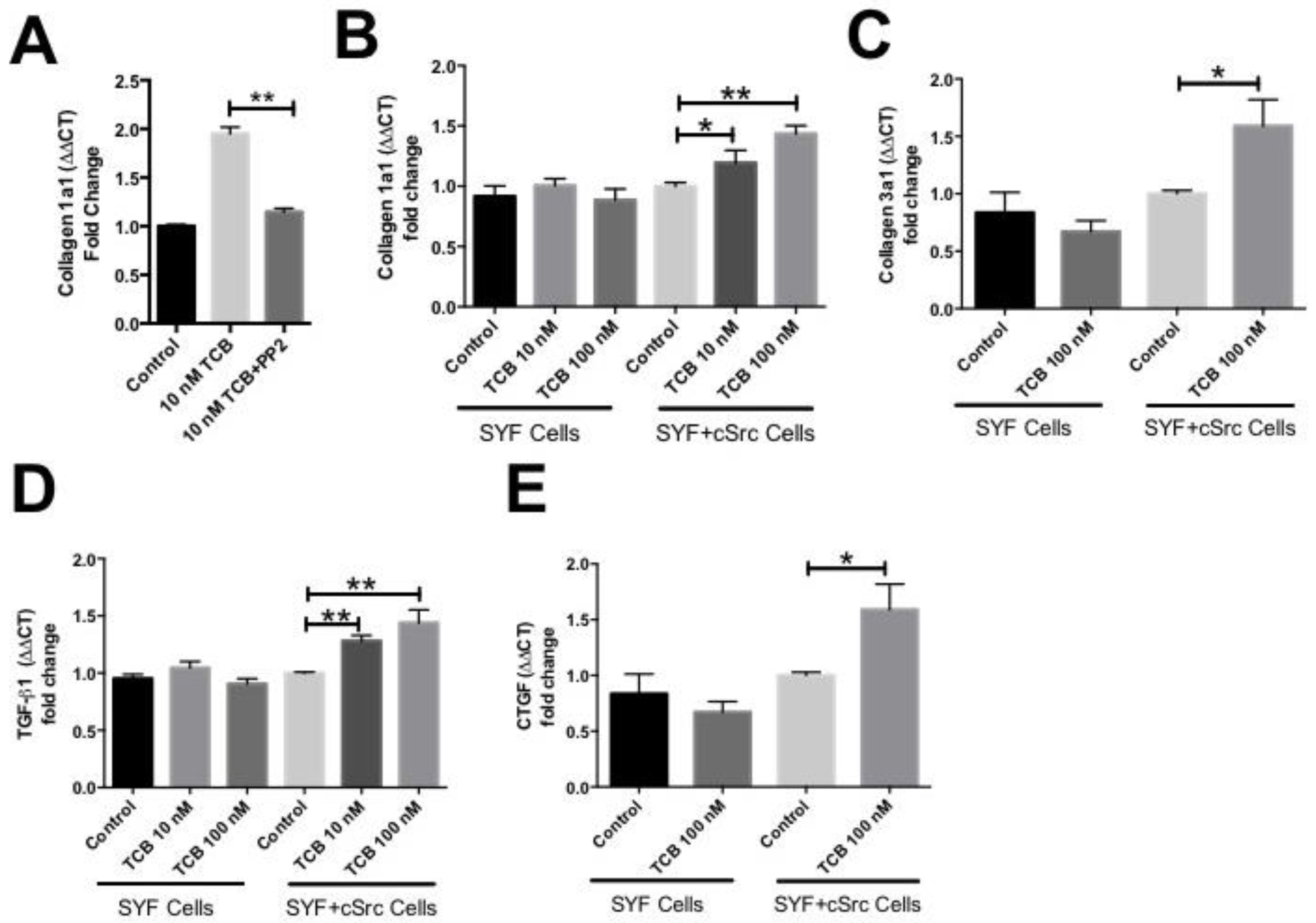
3.2. Pro-Fibrotic Effects of Telocinobufagin Depend on Na+/K+-ATPase Signaling
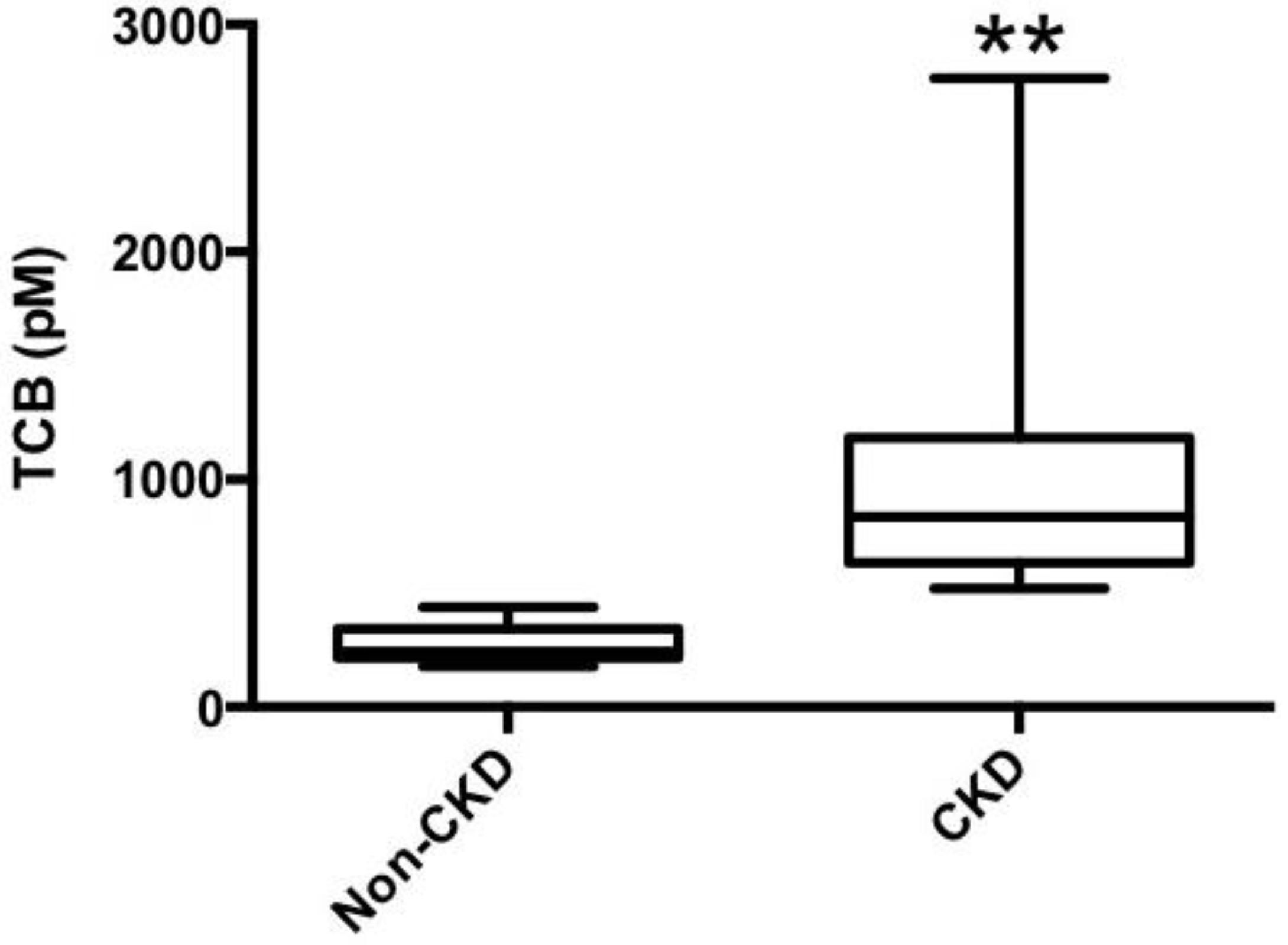
3.3. Telocinobufagin is Elevated in Human Chronic Kidney Disease
4. Discussion
4.1. Mechanisms Linking Cardiotonic Steroids to Renal Dysfunction
4.2. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Damman, K.; Valente, M.A.; Voors, A.A.; O’Connor, C.M.; van Veldhuisen, D.J.; Hillege, H.L. Renal impairment, worsening renal function, and outcome in patients with heart failure: An updated meta-analysis. Eur. Heart J. 2014, 35, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.E.; Butler, J.; Wang, Y.; Abraham, W.T.; O’Connor, C.M.; Gottlieb, S.S.; Loh, E.; Massie, B.M.; Rich, M.W.; Stevenson, L.W.; et al. Incidence, predictors at admission, and impact of worsening renal function among patients hospitalized with heart failure. J. Am. Coll. Cardiol. 2004, 43, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Xie, Z.J. The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim. Biophys. Acta 2010, 1802, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kennedy, D.J.; Yan, Y.; Shapiro, J.I. Reactive Oxygen Species Modulation of Na/K-ATPase Regulates Fibrosis and Renal Proximal Tubular Sodium Handling. Int. J. Nephrol. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, M.; Tian, J.; Liu, L.; Pierre, S.; Liu, J.; Shapiro, J.; Xie, Z.J. Identification of a pool of non-pumping Na/K-ATPase. J. Biol. Chem. 2007, 282, 10585–10593. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, T.; Tian, J.; Xie, J.X.; Zhao, X.; Liu, L.; Shapiro, J.I.; Xie, Z. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J. Biol. Chem. 2009, 284, 21066–21076. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of Src to Na+/K+-ATPase Forms a Functional Signaling Complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Wansapura, A.N.; Lasko, V.; Xie, Z.; Fedorova, O.V.; Bagrov, A.Y.; Lingrel, J.B.; Lorenz, J.N. Marinobufagenin enhances cardiac contractility in mice with ouabain-sensitive alpha1 Na+-K+-ATPase. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1833–H1839. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Askari, A.; Xie, Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [PubMed]
- Liu, L.; Mohammadi, K.; Aynafshar, B.; Wang, H.; Li, D.; Liu, J.; Ivanov, A.V.; Xie, Z.; Askari, A. Role of caveolae in signal-transducing function of cardiac Na+/K+-ATPase. Am. J. Physiol. Cell Physiol. 2003, 284, C1550–C1560. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Cai, T.; Tian, J.; Ivanov, A.V.; Giovannucci, D.R.; Xie, Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol. Biol. Cell 2005, 16, 4034–4045. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Li, X.; Liang, M.; Liu, L.; Xie, J.X.; Ye, Q.; Kometiani, P.; Tillekeratne, M.; Jin, R.; Xie, Z. Changes in sodium pump expression dictate the effects of ouabain on cell growth. J. Biol. Chem. 2009, 284, 14921–14929. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Kometiani, P.; Liu, J.; Li, J.; Shapiro, J.I.; Askari, A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J. Biol. Chem. 1999, 274, 19323–19328. [Google Scholar] [CrossRef] [PubMed]
- Elkareh, J.; Kennedy, D.J.; Yashaswi, B.; Vetteth, S.; Shidyak, A.; Kim, E.G.; Smaili, S.; Periyasamy, S.M.; Hariri, I.M.; Fedorova, L.; et al. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 2007, 49, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Elkareh, J.; Periyasamy, S.M.; Shidyak, A.; Vetteth, S.; Schroeder, J.; Raju, V.; Hariri, I.M.; El-Okdi, N.; Gupta, S.; Fedorova, L.; et al. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli-1: Implications for uremic cardiomyopathy. Am. J. Physiol. Renal Physiol. 2009, 296, F1219–F1226. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, J.; Kennedy, D.J. Regulation of Cardiac Remodeling by Cardiac Na(+)/K(+)-ATPase Isoforms. Front. Physiol. 2016, 7, 382. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Chen, Y.; Huang, W.; Viterna, J.; Liu, J.; Westfall, K.; Tian, J.; Bartlett, D.J.; Tang, W.H.; Xie, Z.; et al. CD36 and Na/K-ATPase-alpha1 form a proinflammatory signaling loop in kidney. Hypertension 2013, 61, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Vetteth, S.; Periyasamy, S.M.; Kanj, M.; Fedorova, L.; Khouri, S.; Kahaleh, M.B.; Xie, Z.; Malhotra, D.; Kolodkin, N.I.; et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006, 47, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Dial, L.D.; Shapiro, J.I. Na/K-ATPase Signaling and the Tradeoff Between Natriuresis and Cardiac Fibrosis. In Cardiomyopathies; Milei, J., Ed.; InTech: Christchurch, New Zealand, 2013. [Google Scholar] [Green Version]
- Kolmakova, E.V.; Haller, S.T.; Kennedy, D.J.; Isachkina, A.N.; Budny, G.V.; Frolova, E.V.; Piecha, G.; Nikitina, E.R.; Malhotra, D.; Fedorova, O.V.; et al. Endogenous cardiotonic steroids in chronic renal failure. Nephrol. Dial. Transplant. 2011, 26, 2912–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorova, L.V.; Raju, V.; El-Okdi, N.; Shidyak, A.; Kennedy, D.J.; Vetteth, S.; Giovannucci, D.R.; Bagrov, A.Y.; Fedorova, O.V.; Shapiro, J.I.; et al. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: Implication of epithelial-to-mesenchymal transition. Am. J. Physiol. Renal Physiol. 2009, 296, F922–F934. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Shrestha, K.; Sheehey, B.; Li, X.S.; Guggilam, A.; Wu, Y.; Finucan, M.; Gabi, A.; Medert, C.M.; Westfall, K.; et al. Elevated Plasma Marinobufagenin, An Endogenous Cardiotonic Steroid, Is Associated with Right Ventricular Dysfunction and Nitrative Stress in Heart Failure. Circ. Heart Fail. 2015, 8, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.A.; Fan, X.; Haller, S.T.; Kennedy, D.J.; Liu, J.; Tian, J. Na/K-ATPase signaling mediates miR-29b-3p regulation and cardiac fibrosis formation in mice with chronic kidney disease. PLoS ONE 2018, 13, e0197688. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Chaudhry, M.; Maxwell, K.; Yan, Y.; Wang, X.; Shah, P.T.; Khawaja, A.A.; Martin, R.; Robinette, T.J.; et al. Attenuation of Na/K-ATPase Mediated Oxidant Amplification with pNaKtide Ameliorates Experimental Uremic Cardiomyopathy. Sci. Rep. 2016, 6, 34592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, T.; Wang, H.; Chen, Y.; Liu, L.; Gunning, W.T.; Quintas, L.E.; Xie, Z.J. Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J. Cell Biol. 2008, 182, 1153–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Li, H.; Xie, Z. Ouabain-induced hypertrophy in cultured cardiac myocytes is accompanied by changes in expression of several late response genes. J. Mol. Cell Cardiol. 1997, 29, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cai, T.; Yang, C.; Turner, D.A.; Giovannucci, D.R.; Xie, Z. Regulation of inositol 1,4,5-trisphosphate receptor-mediated calcium release by the Na/K-ATPase in cultured renal epithelial cells. J. Biol. Chem. 2008, 283, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Touza, N.A.; Pocas, E.S.; Quintas, L.E.; Cunha-Filho, G.; Santos, M.L.; Noel, F. Inhibitory effect of combinations of digoxin and endogenous cardiotonic steroids on Na+/K+-ATPase activity in human kidney membrane preparation. Life Sci. 2011, 88, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Ishii, N.; Nambara, T. Occurrence of bufadienolides in the skin of Bufo viridis Laur. Chem. Pharm. Bull. 1986, 34, 3454–3457. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.K.; Anderson, R.C.; Henderson, F.G. Comparison of cardiac action of bufalin, cinobufotalin, and telocinobufagin with cinobufagin. Proc. Soc. Exp. Biol. Med. 1951, 76, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Schmeda-Hirschmann, G.; Quispe, C.; Theoduloz, C.; de Sousa, P.T., Jr.; Parizotto, C. Antiproliferative activity and new argininyl bufadienolide esters from the “cururu” toad Rhinella (Bufo) schneideri. J. Ethnopharmacol. 2014, 155, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.M.; Lima, D.J.; Debiasi, B.W.; Soares, B.M.; Machado Kda, C.; Noronha Jda, C.; Rodrigues Dde, J.; Sinhorin, A.P.; Pessoa, C.; Vieira, G.M., Jr. Antiproliferative activity of Rhinella marina and Rhaebo guttatus venom extracts from Southern Amazon. Toxicon 2013, 72, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.C.; Zhang, B.J.; Xin, X.L.; Huang, S.S.; Deng, S.; Zhang, H.L.; Shu, X.H.; Diao, Y.P.; Cui, J. Simultaneous quantification of seven major bufadienolides in three traditional Chinese medicinal preparations of chansu by HPLC-DAD. Nat. Prod. Commun. 2009, 4, 179–184. [Google Scholar] [PubMed]
- Komiyama, Y.; Dong, X.H.; Nishimura, N.; Masaki, H.; Yoshika, M.; Masuda, M.; Takahashi, H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin. Biochem. 2005, 38, 36–45. [Google Scholar] [CrossRef] [PubMed]
- James, P.F.; Grupp, I.L.; Grupp, G.; Woo, A.L.; Askew, G.R.; Croyle, M.L.; Walsh, R.A.; Lingrel, J.B. Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol. Cell 1999, 3, 555–563. [Google Scholar] [CrossRef]
- Dostanic, I.; Lorenz, J.N.; Schultz Jel, J.; Grupp, I.L.; Neumann, J.C.; Wani, M.A.; Lingrel, J.B. The alpha2 isoform of Na,K-ATPase mediates ouabain-induced cardiac inotropy in mice. J. Biol. Chem. 2003, 278, 53026–53034. [Google Scholar] [CrossRef] [PubMed]
- Dostanic, I.; Schultz, J.E.J.; Lorenz, J.N.; Lingrel, J.B. The alpha 1 isoform of Na,K-ATPase regulates cardiac contractility and functionally interacts and co-localizes with the Na/Ca exchanger in heart. J. Biol. Chem. 2004, 279, 54053–54061. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Kolodkin, N.I.; Agalakova, N.I.; Namikas, A.R.; Bzhelyansky, A.; St-Louis, J.; Lakatta, E.G.; Bagrov, A.Y. Antibody to marinobufagenin lowers blood pressure in pregnant rats on a high NaCl intake. J. Hypertens. 2005, 23, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Talan, M.I.; Agalakova, N.I.; Lakatta, E.G.; Bagrov, A.Y. Endogenous ligand of alpha1 sodium pump, marinobufagenin, is a novel mediator of sodium chloride—Dependent hypertension. Circulation 2002, 105, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A.; Kellum, J.A.; Haase, M.; Muller, C.; Damman, K.; Murray, P.T.; Cruz, D.; House, A.A.; Schmidt-Ott, K.M.; Vescovo, G.; et al. Pathophysiology of the cardiorenal syndromes: Executive summary from the Eleventh Consensus Conference of the Acute Dialysis Quality Initiative (ADQI). Blood Purif. 2014, 37 (Suppl. 2), 2–13. [Google Scholar] [CrossRef]
- Rosner, M.H.; Ronco, C.; Okusa, M.D. The role of inflammation in the cardio-renal syndrome: A focus on cytokines and inflammatory mediators. Semin. Nephrol. 2012, 32, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Eisenhut, M. Changes in ion transport in inflammatory disease. J. Inflamm. 2006, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, I.; Raviv, S.; Sznajder, J.I. Alveolar epithelium and Na,K-ATPase in acute lung injury. Intens. Care Med. 2007, 33, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kennedy, D.J.; Ramakrishnan, D.P.; Yang, M.; Huang, W.; Li, Z.; Xie, Z.; Chadwick, A.C.; Sahoo, D.; Silverstein, R.L. Oxidized LDL-bound CD36 recruits an Na+/K+-ATPase-Lyn complex in macrophages that promotes atherosclerosis. Sci. Signal. 2015, 8, ra91. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.X.; Zhang, S.; Cui, X.; Zhang, J.; Yu, H.; Khalaf, F.K.; Malhotra, D.; Kennedy, D.J.; Shapiro, J.I.; Tian, J.; et al. Na/K-ATPase/src complex mediates regulation of CD40 in renal parenchyma. Nephrol. Dial. Transplant. 2018, 33, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Song, Y.; An, N.; Zeng, S.; Wang, D.; Yu, L.; Zhu, T.; Zhang, T.; Cui, J.; Zhou, C.; et al. The effects of telocinobufagin isolated from Chan Su on the activation and cytokine secretion of immunocytes in vitro. Fundam. Clin. Pharmacol. 2009, 23, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, A.; Ono, K.; Nishio, R.; Igata, H.; Shioi, T.; Matsui, S.; Furukawa, Y.; Iwasaki, A.; Nose, Y.; Sasayama, S. Modulation of cytokine production and protection against lethal endotoxemia by the cardiac glycoside ouabain. Circulation 1997, 96, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Mascarenhas, S.; Da Silva de Oliveira, A.; Amoedo, N.D.; Affonso-Mitidieri, O.R.; Rumjanek, F.D.; Rumjanek, V.M. Modulation of the immune system by ouabain. Ann. N. Y. Acad. Sci. 2009, 1153, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Chen, W.; Cao, D.; Bian, J.; Gong, F.Y.; Cheng, W.; Cheng, S.; Xu, Q.; Hua, Z.C.; Yin, W. Involvement of Na+, K+-ATPase and its inhibitors in HuR-mediated cytokine mRNA stabilization in lung epithelial cells. Cell. Mol. Life Sci. 2011, 68, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Dan, C.; Jinjun, B.; Zi-Chun, H.; Lin, M.; Wei, C.; Xu, Z.; Ri, Z.; Shun, C.; Wen-Zhu, S.; Qing-Cai, J.; et al. Modulation of TNF-alpha mRNA stability by human antigen R miR181s in sepsis-induced immunoparalysis. EMBO Mol. Med. 2014. [Google Scholar] [CrossRef]
- Berendes, E.; Cullen, P.; Van Aken, H.; Zidek, W.; Erren, M.; Hubschen, M.; Weber, T.; Wirtz, S.; Tepel, M.; Walter, M. Endogenous glycosides in critically ill patients. Crit. Care Med. 2003, 31, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Elkareh, J.; Shidyak, A.; Shapiro, A.P.; Smaili, S.; Mutgi, K.; Gupta, S.; Tian, J.; Morgan, E.; Khouri, S.; et al. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am. J. Physiol. Renal Physiol. 2008, 294, F450–F454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, S.T.; Kennedy, D.J.; Shidyak, A.; Budny, G.V.; Malhotra, D.; Fedorova, O.V.; Shapiro, J.I.; Bagrov, A.Y. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am. J. Hypertens. 2012, 25, 690–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, S.T.; Yan, Y.; Drummond, C.A.; Xie, J.; Tian, J.; Kennedy, D.J.; Shilova, V.Y.; Xie, Z.; Liu, J.; Cooper, C.J.; et al. Rapamycin Attenuates Cardiac Fibrosis in Experimental Uremic Cardiomyopathy by Reducing Marinobufagenin Levels and Inhibiting Downstream Pro-Fibrotic Signaling. J. Am. Heart Assoc. 2016, 5, e004106. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.A.; Hill, M.C.; Shi, H.; Fan, X.; Xie, J.X.; Haller, S.T.; Kennedy, D.J.; Liu, J.; Garrett, M.R.; Xie, Z.; et al. Na/K-ATPase signaling regulates collagen synthesis through microRNA-29b-3p in cardiac fibroblasts. Physiol. Genom. 2016, 48, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Okdi, N.; Smaili, S.; Raju, V.; Shidyak, A.; Gupta, S.; Fedorova, L.; Elkareh, J.; Periyasamy, S.; Shapiro, A.P.; Kahaleh, M.B.; et al. Effects of cardiotonic steroids on dermal collagen synthesis and wound healing. J. Appl. Physiol. 2008, 105, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Shidyak, A.; Periyasamy, S.M.; Haller, S.; Taleb, M.; El-Okdi, N.; Elkareh, J.; Gupta, S.; Gohara, S.; Fedorova, O.V.; et al. Spironolactone attenuates experimental uremic cardiomyopathy by antagonizing marinobufagenin. Hypertension 2009, 54, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Pellieux, C.; Foletti, A.; Peduto, G.; Aubert, J.F.; Nussberger, J.; Beermann, F.; Brunner, H.R.; Pedrazzini, T. Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. J. Clin. Investig. 2001, 108, 1843–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, C.; Shesely, E.G.; Potter, D.L.; Beierwaltes, W.H. Cardiovascular and renal phenotype in mice with one or two renin genes. Hypertension 2004, 43, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Watanabe, Y.; Ohno, Y.; Inoue, T.; Kanno, Y.; Suzuki, H.; Okada, H. Mapping quantitative trait loci for proteinuria-induced renal collagen deposition. Kidney Int. 2008, 73, 1017–1023. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Value |
|---|---|
| Demographics: | |
| Age (years) | 72 ± 11 |
| Male gender, n (%) | 11 (52%) |
| Black, n (%) | 16 (76%) |
| Laboratory data: | |
| TCB (pM) | 1001 ± 110 |
| Creatinine (mg/dL) | 4.1 ± 2.2 |
| Sodium (mEq/L) | 140 ± 3 |
| eGFR (mL/min/1.73 m2) | 25 ± 14 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kennedy, D.J.; Khalaf, F.K.; Sheehy, B.; Weber, M.E.; Agatisa-Boyle, B.; Conic, J.; Hauser, K.; Medert, C.M.; Westfall, K.; Bucur, P.; et al. Telocinobufagin, a Novel Cardiotonic Steroid, Promotes Renal Fibrosis via Na+/K+-ATPase Profibrotic Signaling Pathways. Int. J. Mol. Sci. 2018, 19, 2566. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092566
Kennedy DJ, Khalaf FK, Sheehy B, Weber ME, Agatisa-Boyle B, Conic J, Hauser K, Medert CM, Westfall K, Bucur P, et al. Telocinobufagin, a Novel Cardiotonic Steroid, Promotes Renal Fibrosis via Na+/K+-ATPase Profibrotic Signaling Pathways. International Journal of Molecular Sciences. 2018; 19(9):2566. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092566
Chicago/Turabian StyleKennedy, David J., Fatimah K. Khalaf, Brendan Sheehy, Malory E. Weber, Brendan Agatisa-Boyle, Julijana Conic, Kayla Hauser, Charles M. Medert, Kristen Westfall, Philip Bucur, and et al. 2018. "Telocinobufagin, a Novel Cardiotonic Steroid, Promotes Renal Fibrosis via Na+/K+-ATPase Profibrotic Signaling Pathways" International Journal of Molecular Sciences 19, no. 9: 2566. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092566