Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting

 , ,
, , 

Abstract
:
1. Introduction
2. Molecular Chaperones and Neurodegenerative Diseases
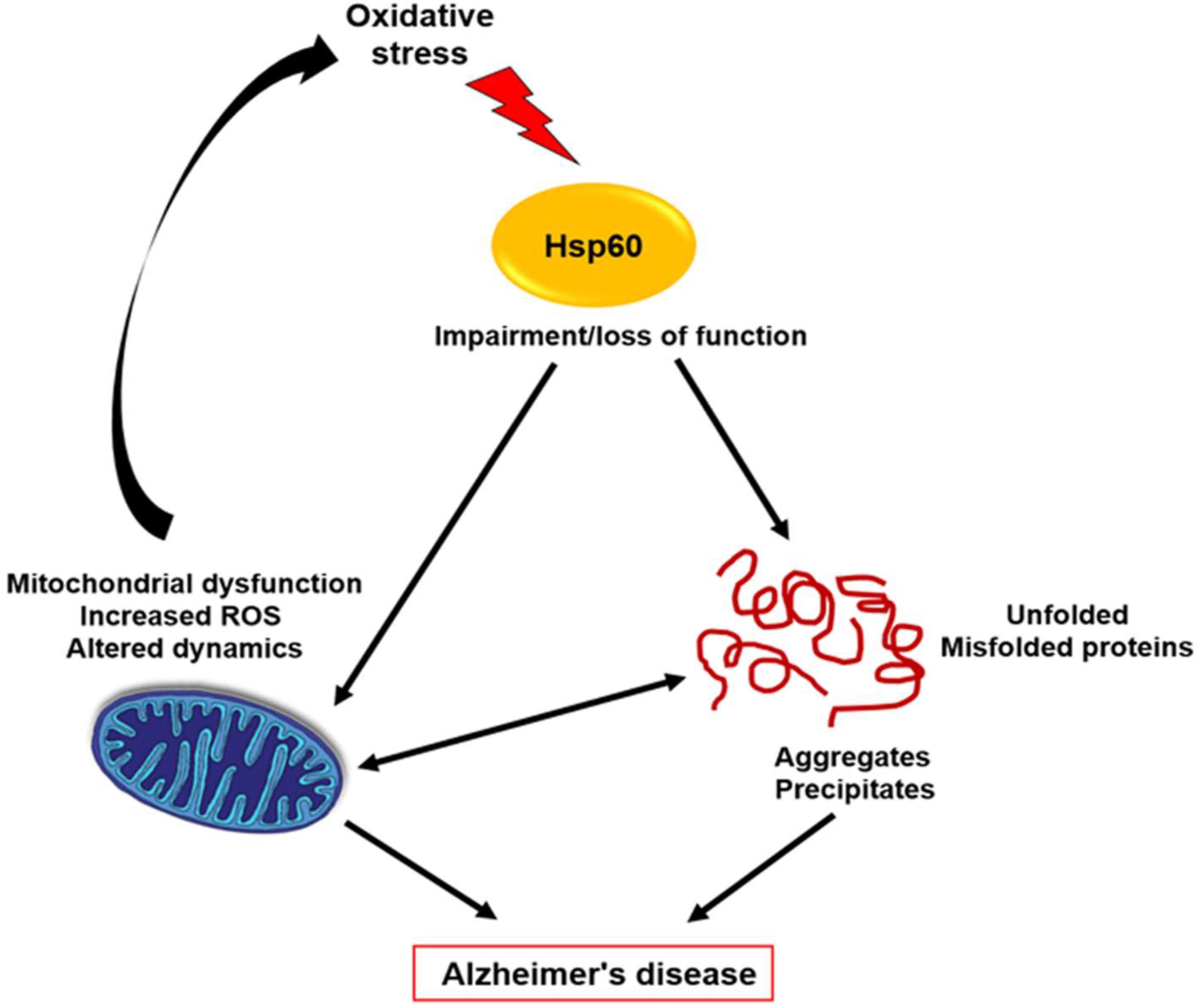
3. Hsp60
3.1. Biological Role in AD
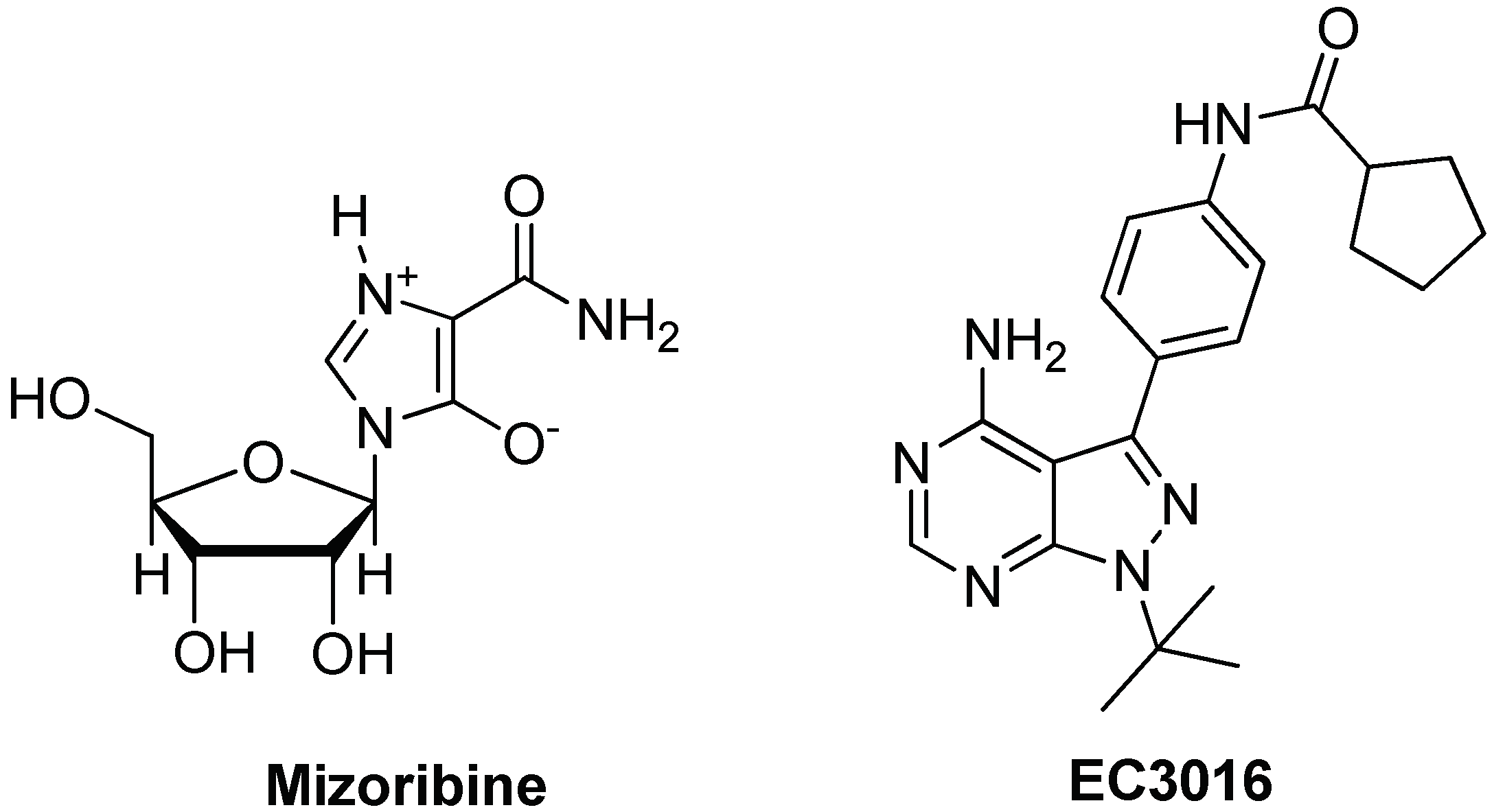
3.2. Targeting and Inhibition
4. Hsp70
4.1. Biological Role in AD
4.2. Targeting and Inhibition
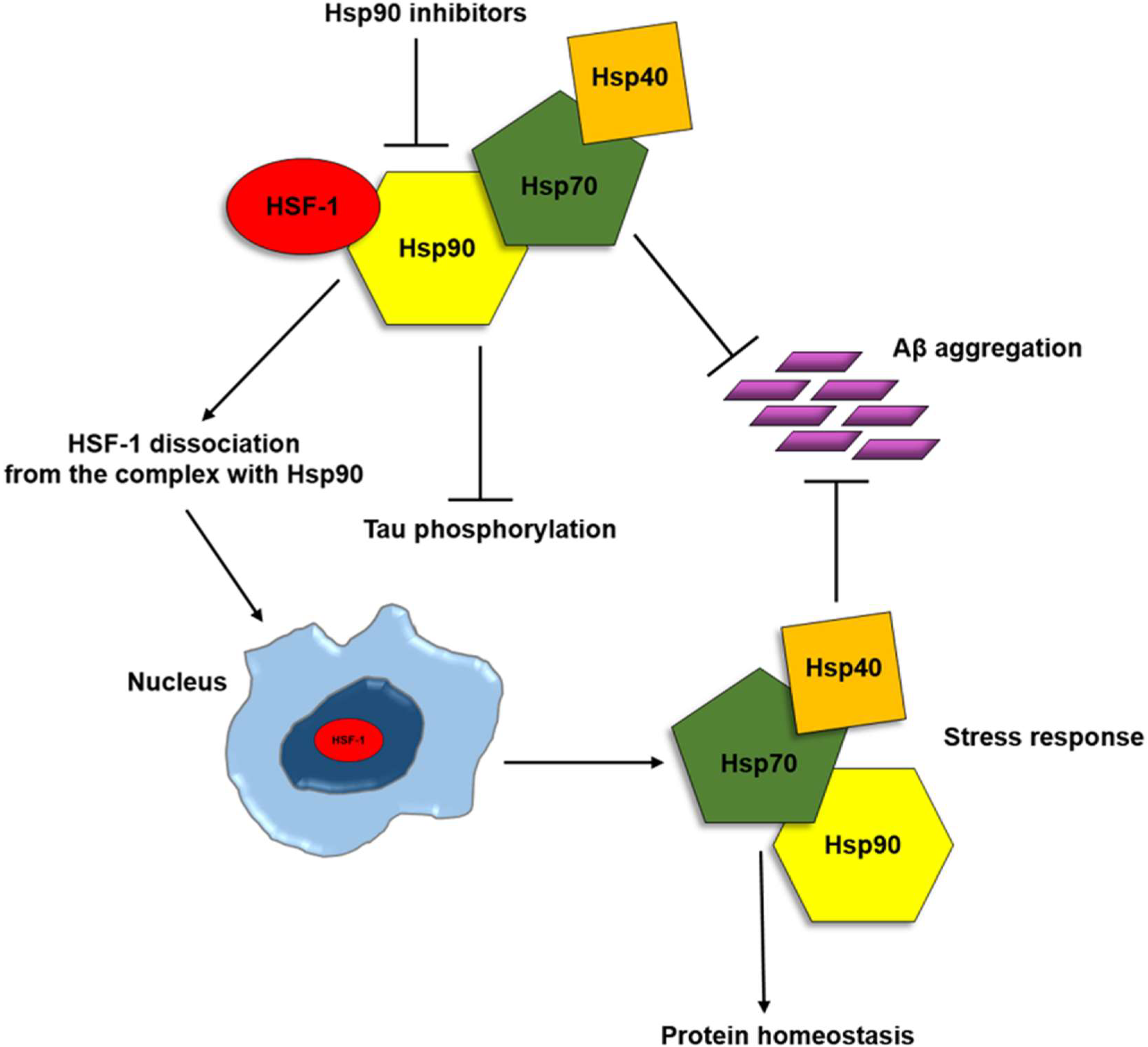
5. Hsp90
5.1. Biological Role in AD
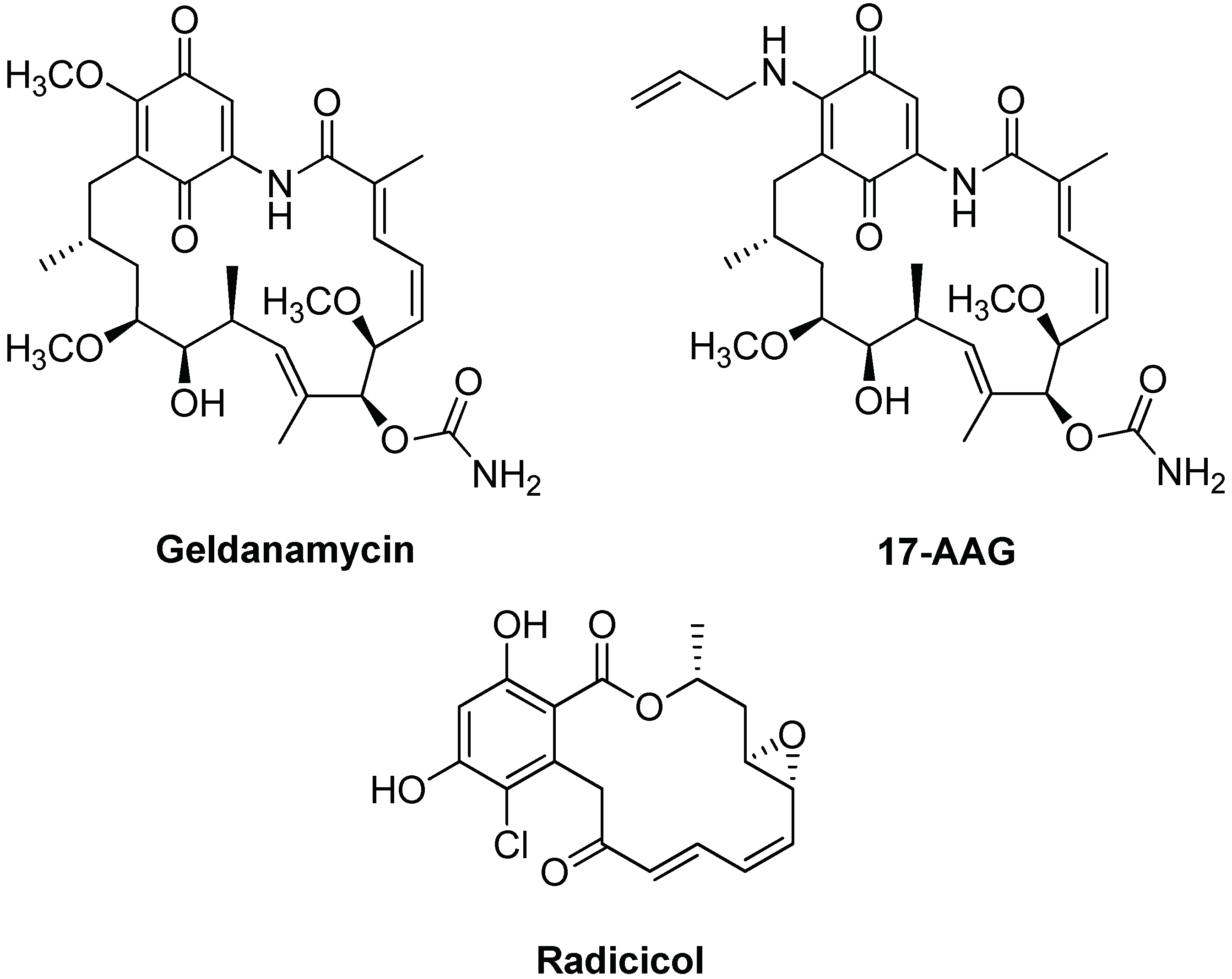
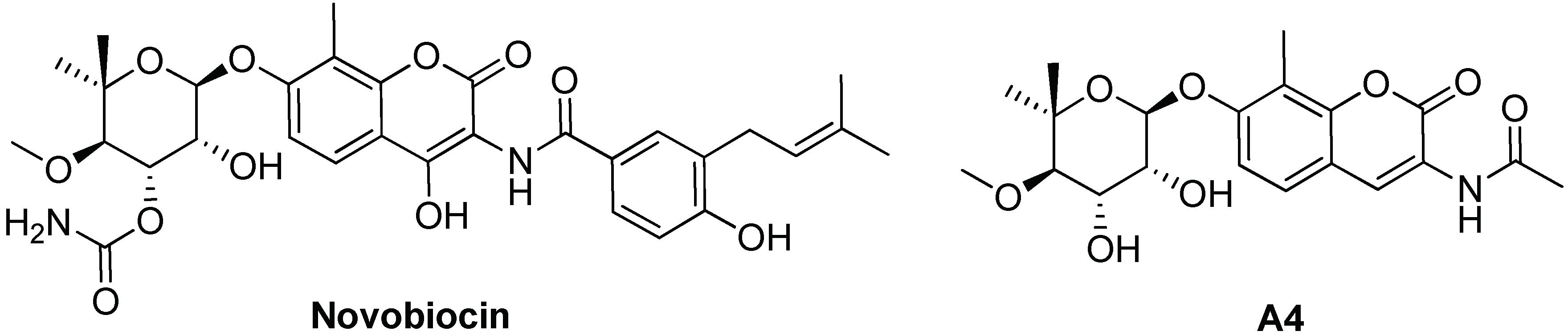
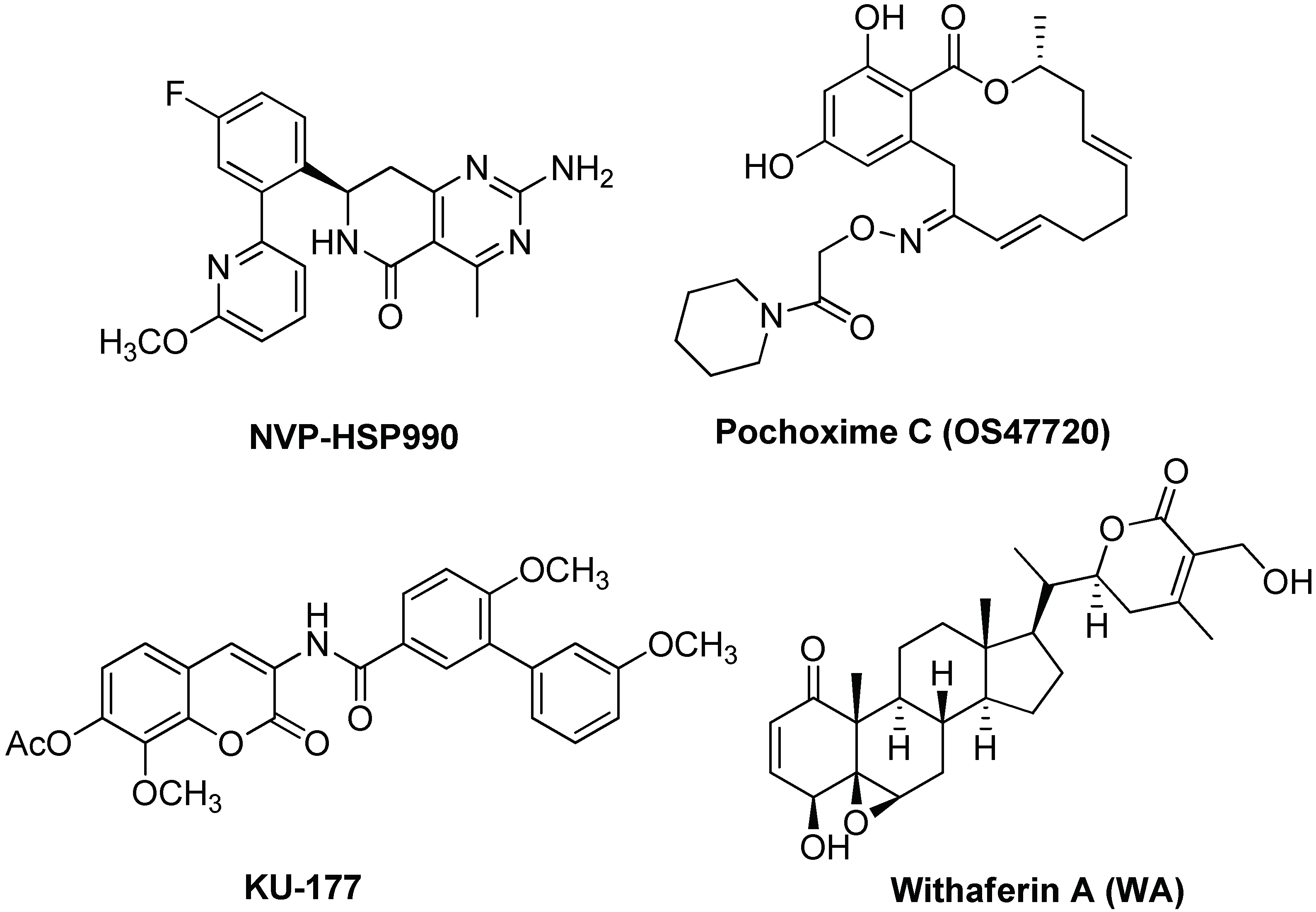
5.2. Targeting and Inhibition
6. Conclusions
Funding
Conflicts of Interest
References
- Hamley, I.W. The amyloid beta peptide: A chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef] [PubMed]
- Parodi, J.; Sepúlveda, F.J.; Roa, J.; Opazo, C.; Inestrosa, N.C.; Aguayo, L.G. β-Amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J. Biol. Chem. 2010, 285, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.E.; Castillo-Carranza, D.L.; Kayed, R. Advances in therapeutics for neurodegenerative tauopathies: Moving toward the specific targeting of the most toxic tau species. ACS Chem. Neurosci. 2014, 5, 752–769. [Google Scholar] [CrossRef] [PubMed]
- Birks, J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, 1, CD005593. [Google Scholar] [CrossRef]
- Kumar, D.; Ganeshpurkar, A.; Kumar, D.; Modi, G.; Gupta, K.S.; Singh, K.S. Secretase inhibitors for the treatment of Alzheimer’s disease: Long road ahead. Eur. J. Med. Chem. 2018, 148, 436–452. [Google Scholar] [CrossRef] [PubMed]
- Martorana, A.; Giacalone, V.; Bonsignore, R.; Pace, A.; Gentile, C.; Pibiri, I.; Buscemi, S.; Lauria, A.; Palumbo Piccionello, A. Heterocyclic scaffolds for the treatment of Alzheimer’s disease. Curr. Pharm. Des. 2016, 22, 3971–3995. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.E.; Kayed, R. Therapeutic approaches targeting pathological tau aggregates. Curr. Pharm. Des. 2016, 22, 4028–4039. [Google Scholar] [CrossRef] [PubMed]
- Battistini, A.; Palumbo Piccionello, A.; Sgarbossa, A.; Vilasi, S.; Ricci, C.; Ghetti, F.; Spinozzi, F.; Marino Gammazza, A.; Giacalone, V.; Martorana, A.; et al. Curcumin-like compounds designed to modify amyloid beta peptide aggregation patterns. RSC Adv. 2017, 7, 31714. [Google Scholar] [CrossRef]
- Mangione, M.R.; Palumbo Piccionello, A.; Marino, C.; Ortore, M.G.; Picone, P.; Vilasi, S.; Di Carlo, M.; Buscemi, S.; Bulone, D.; San Biagio, P.L. Photo-inhibition of Aβ fibrillation mediated by a newly designed fluorinated oxadiazole. RSC Adv. 2015, 5, 16540–16548. [Google Scholar] [CrossRef]
- Yang, X.; Cai, P.; Liu, Q.; Wu, J.; Yin, Y.; Wang, X.; Long, L. Novel 8-hydroxyquinoline derivatives targeting β-amyloid aggregation, metal chelation and oxidative stress against Alzheimer’s disease. Bioorg. Med. Chem. 2018, 26, 3191–3201. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Bonsignore, R.; Barone, G.; Keppler, B.K.; Terenzi, A. Metal ions and metal complexes in Alzheimer’s disease. Curr. Pharm. Des. 2016, 22, 3996–4010. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.; De Macario, E.C. Molecular chaperones and age-related degenerative disorders. Adv. Cell. Aging Gerontol. 2001, 7, 131–162. [Google Scholar] [CrossRef]
- Cappello, F.; Marino Gammazza, A.; Vilasi, S.; Ortore, M.G.; San Biagio, P.L.; Campanella, C.; Pace, A.; Palumbo Piccionello, A.; Taglialatela, G.; De Macario, E.C.; et al. Chaperonotherapy for Alzheimer’s Disease: Focusing on HSP60. In Heat Shock Protein-Based Therapies; Asea, A.A.A., Almasoud, N.N., Krishnan, S., Kaur, P., Eds.; Springer: Cham, Switzerland, 2015; Volume 9, pp. 51–76. [Google Scholar] [CrossRef]
- Manos-Turvey, A.; Brodsky, J.L.; Wipf, P. The Effect of Structure and Mechanism of the Hsp70 Chaperone on the Ability to Identify Chemical Modulators and Therapeutics. Top. Med. Chem. 2016, 19, 81–130. [Google Scholar] [CrossRef]
- Turturici, G.; Sconzo, G.; Geraci, F. Hsp70 and its molecular Role in nervous system diseases. Biochem. Res. Int. 2011, 2011, 618127. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, S.N.; Martin, M.D.; Dickey, C.A. Neurodegeneration and the Heat Shock Protein 70 Machinery: Implications for Therapeutic Development. Curr. Top. Med. Chem. 2016, 16, 2741–2752. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Sabbagh, J.J.; Dickey, C.A. Targeting HSP90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin. Ther. Targets 2014, 18, 1219–1232. [Google Scholar] [CrossRef] [PubMed]
- Inda, C.; Bolaender, A.; Tai, W.; Gandu, S.R.; Koren, J., III. Stressing out Hsp 90 in Neurotoxic Proteinopathies. Curr. Top. Med. Chem. 2016, 16, 2829–2838. [Google Scholar] [CrossRef] [PubMed]
- Taldone, T.; Ochiana, S.O.; Patel, P.D.; Chiosis, G. Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol. Sci. 2014, 35, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino Gammazza, A.; Caruso Bavisotto, C.; Barone, R.; Macario, E.C.; Macario, A.J. Alzheimer’s disease and Molecular Chaperones: Current Knowledge and the Future of Chaperonotherapy. Curr. Pharm. Des. 2016, 22, 4040–4049. [Google Scholar] [CrossRef] [PubMed]
- Cappello, F.; Marino Gammazza, A.; Palumbo Piccionello, A.; Campanella, C.; Pace, A.; De Macario, E.C.; Macario, A.J. Hsp60 chaperonopathies and chaperonotherapy: Targets and agents. Expert Opin. Ther. Targets 2014, 18, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Pace, A.; Barone, G.; Lauria, A.; Martorana, A.; Palumbo Piccionello, A.; Pierro, P.; Terenzi, A.; Almerico, A.M.; Buscemi, S.; Campanella, C.; et al. Hsp60, a novel target for antitumor therapy: Structure-function features and prospective drugs design. Curr. Pharm. Des. 2013, 19, 2757–2764. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approch. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2016, 38, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.L.; De Macario, E.C. Chaperonopathies and chaperonotherapy. FEBS Lett. 2007, 581, 3681–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanella, C.; D’Anneo, A.; Marino Gammazza, A.; Caruso Bavisotto, C.; Barone, R.; Emanuele, S.; Lo Cascio, F.; Mocciaro, E.; Fais, S.; De Macario, E.C.; et al. The histone deacetylase inhibitor SAHA induces HSP60 nitration and its extracellular release by exosomal vesicles in human lung-derived carcinoma cells. Oncotarget 2016, 7, 28849–28867. [Google Scholar] [CrossRef] [PubMed]
- Caruso Bavisotto, C.; Nikolic, D.; Marino Gammazza, A.; Barone, R.; Lo Cascio, F.; Mocciaro, E.; Zummo, G.; De Macario, E.C.; Macario, A.J.; Cappello, F.; et al. The dissociation of the Hsp60/pro-Caspase-3- complex by bis (pyrydil)oxadiazole complex (CubipyOXA) leads to cell death in NCl-H292 cancer cells. J. Inorg. Biochem. 2017, 170, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Vilasi, S.; Bulone, D.; Caruso Bavisotto, C.; Campanella, C.; Marino Gammazza, A.; San Biagio, P.L.; Cappello, F.; De Macario, E.C.; Macario, A.J.L. Chaperonin of group I: Oligomeric spectrum and biochemical and biological implications. Front. Mol. Biosc. 2018, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.L.; De Macario, E.C. Molecular chaperones: Multiple functions, pathologies, and potential applications. Front. Biosci. 2007, 12, 2588–2600. [Google Scholar] [CrossRef] [PubMed]
- Rappa, F.; Sciume, C.; Lo Bello, M.; Bavisotto Caruso, C.; Marino Gammazza, A.; Barone, R.; Campanella, C.; David, S.; Carini, F.; Zarcone, F.; et al. Comparative analysis of hsp10 and hsp90 expression in healthy mucosa and adenocarcinoma of the large bowel. Anticancer Res. 2014, 34, 4153–4159. [Google Scholar] [PubMed]
- Maiti, P.; Manna, J.; Veleri, S.; Frautschy, S. Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. Biomed. Res. Int. 2014, 2014, 495091. [Google Scholar] [CrossRef] [PubMed]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopez, J.; Choy, W.Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 cheperone machinery in neurodegenerative diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef] [PubMed]
- Blum, D.; Chern, Y.; Domenici, M.R.; Buée, L.; Lin, C.Y.; Rea, W.; Ferré, S.; Popoli, P. The Role of Adenosine Tone and Adenosine Receptors in Huntington’s Disease. J. Caffeine Adenosine Res. 2018, 8, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, A.K.; Jarosz, D.F. More than Just a Phase: Prions at the Crossroads of Epigenetic Inheritance and Evolutionary Change. J. Mol. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, Z.; Lang, J.; Perrett, R.M.; Elschami, M.; Hurry, M.E.; Kim, H.T.; Mazaraki, D.; Szabo, A.; Kessler, B.M.; Goldberg, A.L.; et al. Deubiquitinase Usp8 regulates α-synuclein clearance and modifies its toxicity in Lewy body disease. Proc. Natl. Acad. Sci. USA 2016, 113, E4688–E4697. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Campanella, C.; Zummo, G.; Cappello, F. Mitochondrial chaperones in cancer: From molecular biology to clinical diagnostics. Cancer Biol. Ther. 2006, 5, 714–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul, H.M.; Butterfield, D.A. Involvement of PI3K/PKG/ERK1/2 signaling pathways in cortical neurons to trigger protection by co-treatment of acetyl-l-carnitine and α-lipoic acid against HNE-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. Free Radical Biomol. 2007, 42, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Simon, A. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meriin, A.B.; Narayanan, A.; Meng, L.; Alexandrov, I.; Varelas, X.; Cissé, I.I.; Sherman, M.Y. Hsp70-Bag3 complex is a hub for proteotoxicity-induced signaling that controls protein aggregation. Proc. Natl. Acad. Sci. USA 2018, 115, E7043–E7052. [Google Scholar] [CrossRef] [PubMed]
- Walls, K.C.; Coskun, P.; Gallegos-Perez, J.L.; Zadourian, N.; Freude, K.; Rasool, S.; Blurton-Jones, M.; Green, K.N.; LaFerla, F.M. Swedish Alzheimer mutation induces 877 mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein 878 (APP) and beta-amyloid. J. Biol. Chem. 2012, 287, 30317–30327. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Ramachandra, N.B.; Bowes, T.; Singh, B. Unusual cellular disposition of the mitochondrial molecular chaperones Hsp60, Hsp70 and Hsp10. Novartis Found. Symp. 2008, 291, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.R.; Le, W.D. Collective roles of molecular chaperones in protein degradation pathways associated with neurodegenerative diseases. Curr. Pharm. Biotechnol. 2010, 11, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Merendino, A.M.; Bucchieri, F.; Campanella, C.; Marcianò, V.; Ribbene, A.; David, S.; Zummo, G.; Burgio, G.; Corona, D.F.; De Macario, C.E.; et al. Hsp60 is actively secreted by human tumor cells. PLoS ONE 2010, 5, e9247. [Google Scholar] [CrossRef] [PubMed]
- Campanella, C.; Bucchieri, F.; Merendino, A.M.; Fucarino, A.; Burgio, G.; Corona, D.F.; Barbieri, G.; David, S.; Farina, F.; Zummo, G.; et al. The odyssey of hsp60 from tumor cells to other destinations includes plasma membrane-associated stages and Golgi and exosomal protein-trafficking modalities. PLoS ONE 2012, 7, e42008. [Google Scholar] [CrossRef] [PubMed]
- Campanella, C.; Rappa, F.; Sciumè, C.; Marino Gammazza, A.; Barone, R.; Bucchieri, F.; David, S.; Curcurù, G.; Caruso Bavisotto, C.; Pitruzzella, A.; et al. Heat shock protein 60 levels in tissue and circulating exosomes in human large bowel cancer before and after ablative surgery. Cancer 2015, 121, 3230–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso Bavisotto, C.; Marino Gammazza, A.; Rappa, F.; Fucarino, A.; Pitruzzella, A.; David, S.; Campanella, C. Exosomes: Can doctors still ignore their existence? EuroMediterr. Biomed. J. 2013, 8, 137–139. [Google Scholar] [CrossRef]
- Chandra, D.; Choy, G.; Tang, D.G. Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: Evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. J. Biol. Chem. 2007, 282, 31289–31301. [Google Scholar] [CrossRef] [PubMed]
- Campanella, C.; Bucchieri, F.; Ardizzone, N.M.; Marino Gammazza, A.; Montalbano, A.; Ribbene, A.; Di Felice, V.; Bellafiore, M.; David, S.; Rappa, F.; et al. Upon oxidative stress, the antiapoptotic Hsp60/procaspase-3 complex persists in mucoepidermoid carcinoma cells. Eur. J. Histochem. 2008, 52, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.C.; Dohi, T.; Kang, B.H.; Altieri, D.C. Hsp60 regulation of tumor cell apoptosis. J. Biol. Chem. 2008, 283, 5188–5194. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Knowlton, A.A. HSP60, Bax, apoptosis and the heart. J. Cell. Mol. Med. 2005, 9, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Xanthoudakis, S.; Roy, S.; Rasper, D.; Hennessey, T.; Aubin, Y.; Cassady, R.; Tawa, P.; Ruel, R.; Rosen, A.; Nicholson, D.W. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J. 1999, 18, 2049–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorska, M.; Marino Gammazza, A.; Zmijewski, M.A.; Campanella, C.; Cappello, F.; Wasiewicz, T.; Kuban-Jankowska, A.; Daca, A.; Sielicka, A.; Popowska, U.; et al. Geldanamycin-induced osteosarcoma cell death is associated with hyperacetylation and loss of mitochondrial pool of heat shock protein 60 (hsp60). PLoS ONE 2013, 8, e71135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zheng, J.; Xu, Y.; Zhang, X. Paraquat-induced inflammatory response of microglia through HSP60/TLR4 signaling. Hum. Exp. Toxicol. 2018, 1, 960327118758152. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Yamashita, T.; Nagano, K.; Otani, M.; Maekura, K.; Kamada, H.; Tsunoda, S.; Tsutsumi, Y.; Tomiyama, T.; Mori, H.; et al. Proteomic analysis of the hippocampus in Alzheimer’s disease model mice by using two-dimensional fluorescence difference in gel electrophoresis. Neurosci. Lett. 2013, 534, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Li, Y.; Hou, X.; Miao, Z.; Wang, Y. Neuroprotective effect of heat shock protein 60 on matrine-suppressed microglial activation. Exp. Ther. Med. 2017, 14, 1832–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Hong, J.S. Role of microglia in inflammation-mediated neurodegenerative disease: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cappello, F.; De Macario, C.E.; Marino Gammazza, A.; Bonaventura, G.; Carini, F.; Czarnecka, A.M.; Farina, F.; Zummo, G.; Macario, A.J.L. Hsp60 and human aging: Les liaisons dangereuses. Front. Biosci. 2013, 18, 626–637. [Google Scholar] [CrossRef]
- Wojsiat, J.; Prandelli, C.; Laskowska-Kaszub, K.; Martín-Requero, A.; Wojda, U. Oxidative Stress and Aberrant Cell Cycle in Alzheimer’s Disease Lymphocytes: Diagnostic Prospects. J. Alzheimers Dis. 2015, 46, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, A.M.; Pulugulla, S.H.; Pant, D.B.; Posimo, J.M.; Crum, T.S.; Leak, R.K. Impact of aging on heat shock protein expression in the substantia nigra and striatum of the female rat. Cell Tissue Res. 2014, 357, 43–54. [Google Scholar] [CrossRef]
- Bross, P.; Magnoni, R.; Bie, A.S. Molecular chaperone disorders: Defective Hsp60 in neurodegeneration. Curr. Top. Med. Chem. 2012, 12, 2491–2503. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Aβ toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Veereshwarayya, V.; Kumar, P.; Rosen, K.M.; Mestril, R.; Querfurth, H.W. Differential effects of mitochondrial heat shock protein 60 and related molecular chaperones to prevent intracellular beta-amyloid-induced inhibition of complex IV and limit apoptosis. J. Biol. Chem. 2006, 281, 29468–29478. [Google Scholar] [CrossRef] [PubMed]
- Mangione, M.R.; Vilasi, S.; Marino, C.; Librizzi, F.; Canale, C.; Spigolon, D.; Bucchieri, F.; Fucarino, A.; Passantino, R.; Cappello, F.; et al. Hsp60, amateur chaperone in amyloid-beta fibrillogenesis. Biochim. Biophys. Acta 2016, 1860, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Nemirovsky, A.; Fisher, Y.; Baron, R.; Cohen, I.R.; Monsonego, A. Amyloid beta-HSP60 peptide conjugate vaccine treats a mouse model of Alzheimer’s disease. Vaccine 2011, 29, 4043–4050. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, Y.; Osada, H. High-throughput screening identifies small molecule inhibitors of molecular chaperones. Curr. Pharm. Des. 2013, 19, 473–492. [Google Scholar] [CrossRef] [PubMed]
- Qianli, M.; Bingbing, X.L.; Xiangshu, X. Toward Developing Chemical Modulators of Hsp60 as potential Therapeutics. Front. Mol. Biosci. 2018, 5, 35. [Google Scholar] [CrossRef]
- Nisemblat, S.; Parnas, A.; Yaniv, O.; Azem, A.; Frolow, F. Crystallization and structure determination of a symmetrical ‘football’ complex of the mammalian mitochondrial Hsp60-Hsp10 chaperonins. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Nisemblat, S.; Parnas, A.; Yaniv, O.; Azem, A. Crystal structure of the human mitochondrial chaperonin symmetrical football complex. Proc. Natl. Acad. Sci. USA 2015, 112, 6044–6049. [Google Scholar] [CrossRef] [PubMed]
- Ishida, R.; Okamoto, T.; Motojima, F.; Kubota, H.; Takahashi, H.; Tanabe, M.; Oka, T.; Kitamura, A.; Kinjo, M.; Yoshida, M.; et al. Physicochemical properties of the mammalian molecular chaperone HSP60. Int. J. Mol. Sci. 2018, 19, 489. [Google Scholar] [CrossRef] [PubMed]
- Vilasi, S.; Carrotta, R.; Mangione, M.R.; Campanella, C.; Librizzi, F.; Randazzo, L.; Martorana, V.; Marino Gammazza, A.; Ortore, M.G.; Vilasi, A.; et al. Human Hsp60 with its mitochondrial import signal occurs in solution as heptamers and tetradecamers remarkably stable over a wide range of concentrations. PLoS ONE 2014, 9, e97657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinello, A.; Ortore, M.G.; Spinozzi, F.; Ricci, C.; Barone, G.; Marino Gammazza, A.; Palumbo Piccionello, A. Quaternary structures of GroEL and naïve-Hsp60 chaperonins in solution: A combined SAXS-MD study. RCS Adv. 2015, 5, 49871–49879. [Google Scholar] [CrossRef]
- Itoh, H.; Komatsuda, A.; Wakui, H.; Miura, A.B.; Tashima, Y. Mammalian HSP60 is a major target for an immunosuppressant mizoribine. J. Biol. Chem. 1999, 274, 35147–35151. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.; Farr, G.W.; Fenton, W.A.; Johnson, S.M.; Horwich, A.L. Requirement for binding multiple ATPs to convert a GroEL ring to the folding-active state. Proc. Natl. Acad. Sci. USA 2008, 105, 19205–19210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanabe, M.; Ishida, R.; Izuhara, F.; Komatsuda, A.; Wakui, H.; Sawada, K.; Otaka, M.; Nakamura, N.; Itoh, H. The ATPase activity of molecular chaperone HSP60 is inhibited by immunosuppressant mizoribine. Am. J. Mol. Biol. 2012, 2, 93–102. [Google Scholar] [CrossRef]
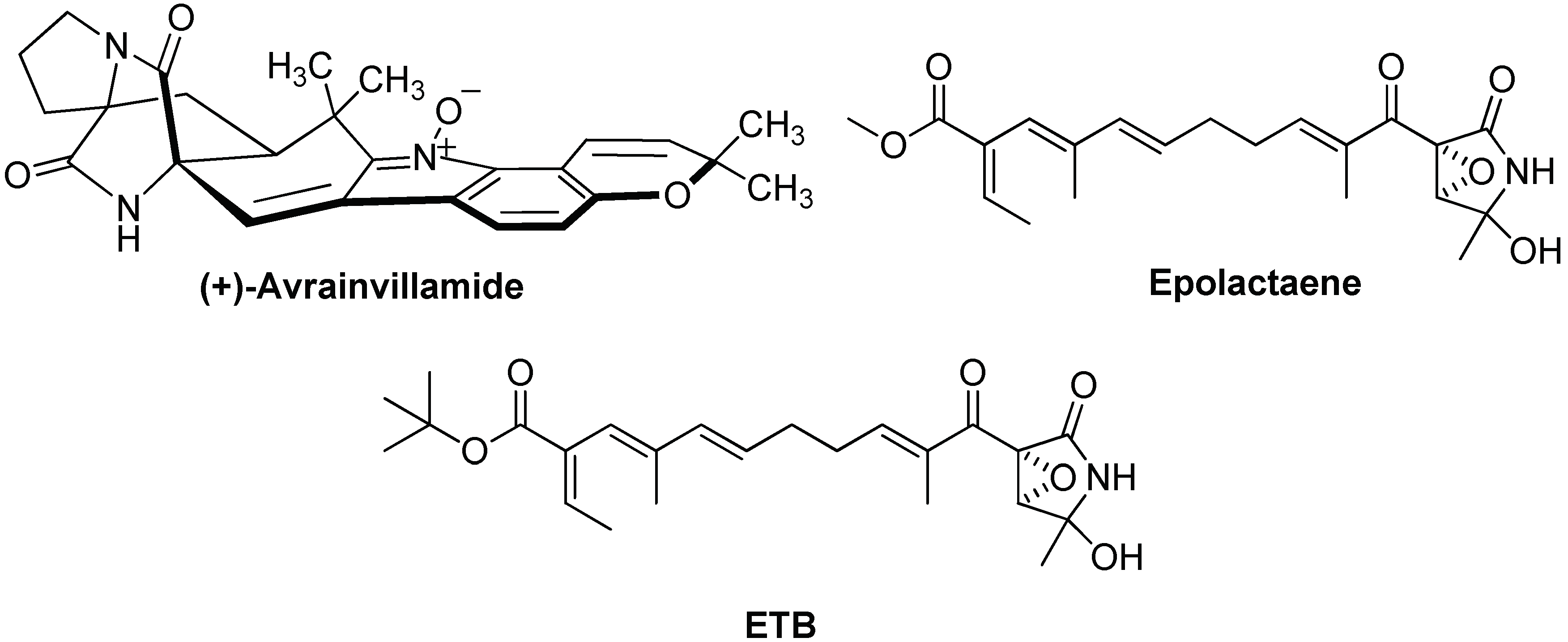
- Wulff, J.E.; Herzon, S.B.; Siegrist, R.; Myers, A.G. Evidence for the rapid conversion of Stephacidin b into the electrophilic monomer avrainvillamide in cell culture. J. Am. Chem. Soc. 2007, 129, 4898–4899. [Google Scholar] [CrossRef] [PubMed]
- Nagumo, Y.; Kakeya, H.; Shoji, M.; Hayashi, Y.; Dohmae, N.; Osada, H. Epolactaene binds human Hsp60 Cys442 resulting in the inhibition of chaperone activity. Biochem. J. 2005, 387, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wang, L.; Jiang, H.; Murchie, A.I. Targeting mitochondrial transcription in fission yeast with ETB, an inhibitor of HSP60, the chaperone that binds to the mitochondrial transcription factor Mtf1. Genes Cells 2012, 17, 122–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagumo, Y.; Kakeya, H.; Yamaguchi, J.; Uno, T.; Shoji, M.; Hayashi, Y.; Osada, H. Structure-activity relationships of epolactaene derivatives: Structural requirements for inhibition of Hsp60 chaperone activity. Bioorg. Med. Chem. Lett. 2004, 14, 4425–4429. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Barone, G.; Cappello, F.; Pace, A.; Buscemi, S.; Palumbo Piccionello, A. The Binding Mechanism of Epolactaene to Hsp60 Unveiled by in Silico Modelling. ChemistrySelect 2016, 4, 759–765. [Google Scholar] [CrossRef]
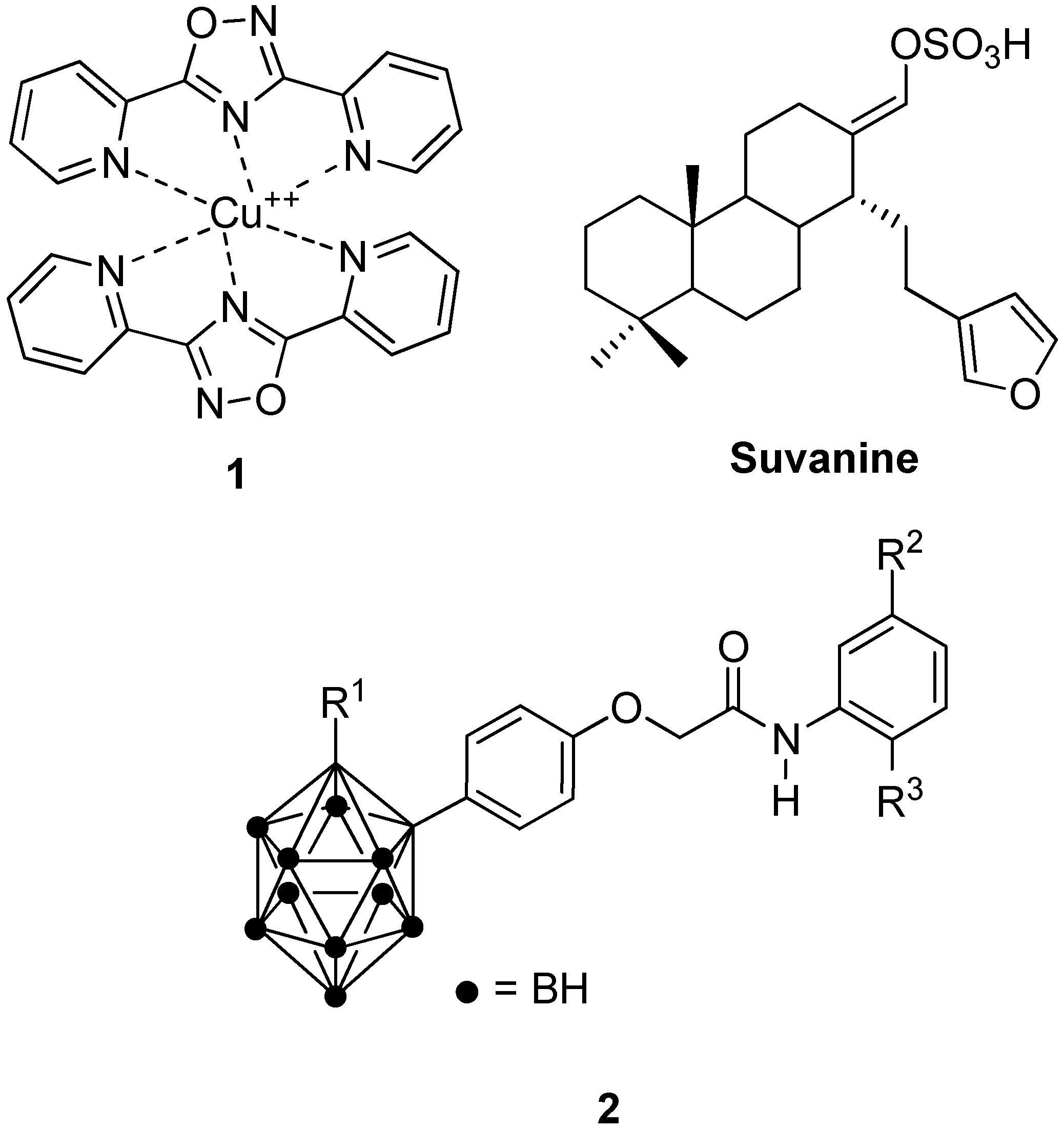
- Terenzi, A.; Barone, G.; Palumbo Piccionello, A.; Giorgi, G.; Portanova, P.; Calvaruso, G.; Buscemi, S.; Vivona, N.; Pace, A. Synthesis, characterization, cellular uptake and interaction with native DNA of a bis (pyridyl)-1,2,4-oxadiazole copper(II) complex. Dalton Trans. 2010, 39, 9140–9145. [Google Scholar] [CrossRef] [PubMed]
- Cassiano, C.; Monti, M.C.; Festa, C.; Zampella, A.; Riccio, R.; Casapullo, A. Chemical proteomics reveals heat shock protein 60 to be the main cellular target of the marine bioactive sesterterpene Suvanine. ChemBioChem 2012, 13, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.S.; Shimizu, K.; Minegishi, H.; Nakamura, H. Identification of HSP60 as a primary target of o-carboranylphenoxyacetanilide, an HIF1alpha inhibitor. J. Am. Chem. Soc. 2010, 132, 11870–11871. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.S.; Shimizu, K.; Minegishi, H.; Nakamura, H. Identification of heat shock protein 60 as the regulator of the hypoxia-inducible factor subunit HIF-1. Pure Appl. Chem. 2012, 84, 2325–2337. [Google Scholar] [CrossRef]
- Nakamura, H.; Yasui, Y.; Maruyama, M.; Minegishi, H.; Ban, H.S.; Sato, S. Development of hypoxia-inducible factor (HIF)-1a inhibitors: Effect of ortho-carborane substituents on HIF transcriptional activity under hypoxia. Bioorg. Med. Chem. Lett. 2013, 23, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.E. The HSP70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jores, T.; Lawatscheck, J.; Beke, V.; Franz-Wachtel, M.; Yunoki, K.; Fitzgerald, J.C.; Macek, B.; Endo, T.; Kalbacher, H.; Buchner, J.; et al. Cytosolic Hsp70 and Hsp40 chaperones enable the biogenesis of mitochondrial β-barrel proteins. J. Cell. Biol. 2018, 217, jcb.201712029. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Hosono, M.; Ogawa, Y.; Inage, K.; Sugawara, S.; Nitta, K. Downregulation of Hsp70 inhibits apoptosis induced by sialic acid-binding lectin (leczyme). Oncol. Rep. 2014, 31, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Alani, B.; Salehi, R.; Sadeghi, P.; Khodagholi, F.; Digaleh, H.; Jabbarzadeh-Tabrizi, S.; Zare, M.; Korbekandi, H. Silencing of Hsp70 intensifies 6-OHDA-induced apoptosis and Hsp90 upregulation in PC12 cells. J. Mol. Neurosci. 2015, 55, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Nadin, S.B.; Sottile, M.L.; Montt-Guevara, M.M.; Gauna, G.V.; Daguerre, P.; Leuzzi, M.; Gago, F.E.; Ibarra, J.; Cuello-Carrión, F.D.; Ciocca, D.R.; et al. Prognostic implication of HSPA (HSP70) in breast cancer patients treated with neoadjuvant anthracycline-based chemotherapy. Cell Stress Chaperones 2014, 19, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Jagadish, N.; Agarwal, S.; Gupta, N.; Fatima, R.; Devi, S.; Kumar, V.; Suri, V.; Kumar, R.; Suri, V.; Sadasukhi, T.C.; et al. Heat shock protein 70-2 (HSP70-2) overexpression in breast cancer. J. Exp. Clin. Cancer Res. 2016, 35, 150. [Google Scholar] [CrossRef] [PubMed]
- Campanella, C.; Caruso Bavisotto, C.; Marino Gammazza, A.; Nikolic, D.; Rappa, F.; David, S.; Cappello, F.; Bucchieri, F.; Fais, S. Exosomal Heat Shock Proteins as New Players in Tumour Cell-to-Cell Communication. J. Circ. Biomark. 2014, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Bausero, M.A.; Gastpar, R.; Multhoff, G.; Asea, A. Alternative mechanism by which IFN-gamma enhances tumor recognition: Active release of heat shock protein 72. J. Immunol. 2005, 175, 2900–2912. [Google Scholar] [CrossRef] [PubMed]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef] [PubMed]
- Cappello, F.; Logozzi, M.; Campanella, C.; Caruso Bavisotto, C.; Marcilla, A.; Properzi, F.; Fais, S. Exosome levels in human body fluids: A tumor marker by themselves? Eur. J. Pharm. Sci. 2017, 96, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.R.; Chen, S. Suppression of Alzheimer’s disease-related phenotypes by the heat shock protein 70 inducer, geranylgeranylacetone, in APP/PS1 transgenic mice via the ERK/p38 MAPK signaling pathway. Exp. Ther. Med. 2017, 14, 5267–5274. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Murao, N.; Namba, T.; Takehara, M.; Adachi, H.; Katsuno, M.; Sobue, G.; Matsushima, T.; Suzuki, T.; Mizushima, T. Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J. Neurosci. 2011, 31, 5225–5234. [Google Scholar] [CrossRef] [PubMed]
- Koren, J.; Jinwal, U.K.; Lee, D.C.; Jones, J.R.; Shults, C.L.; Johnson, A.G.; Anderson, L.J.; Dickey, C.A. Chaperone signalling complexes in Alzheimer’s disease. J. Cell. Mol. Med. 2009, 13, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.G.; Wisen, S.; Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef] [PubMed]
- De Mena, L.; Chhangani, D.; Fernandez-Funez, P.; Rincon-Limas, D.E. secHsp70 as a tool to approach amyloid-β42 and other extracellular amyloids. Fly 2017, 11, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repalli, J.; Meruelo, D. Screening strategies to identify HSP70 modulators to treat Alzheimer’s disease. Drug Des. Dev. Ther. 2015, 9, 321–331. [Google Scholar] [CrossRef] [PubMed]
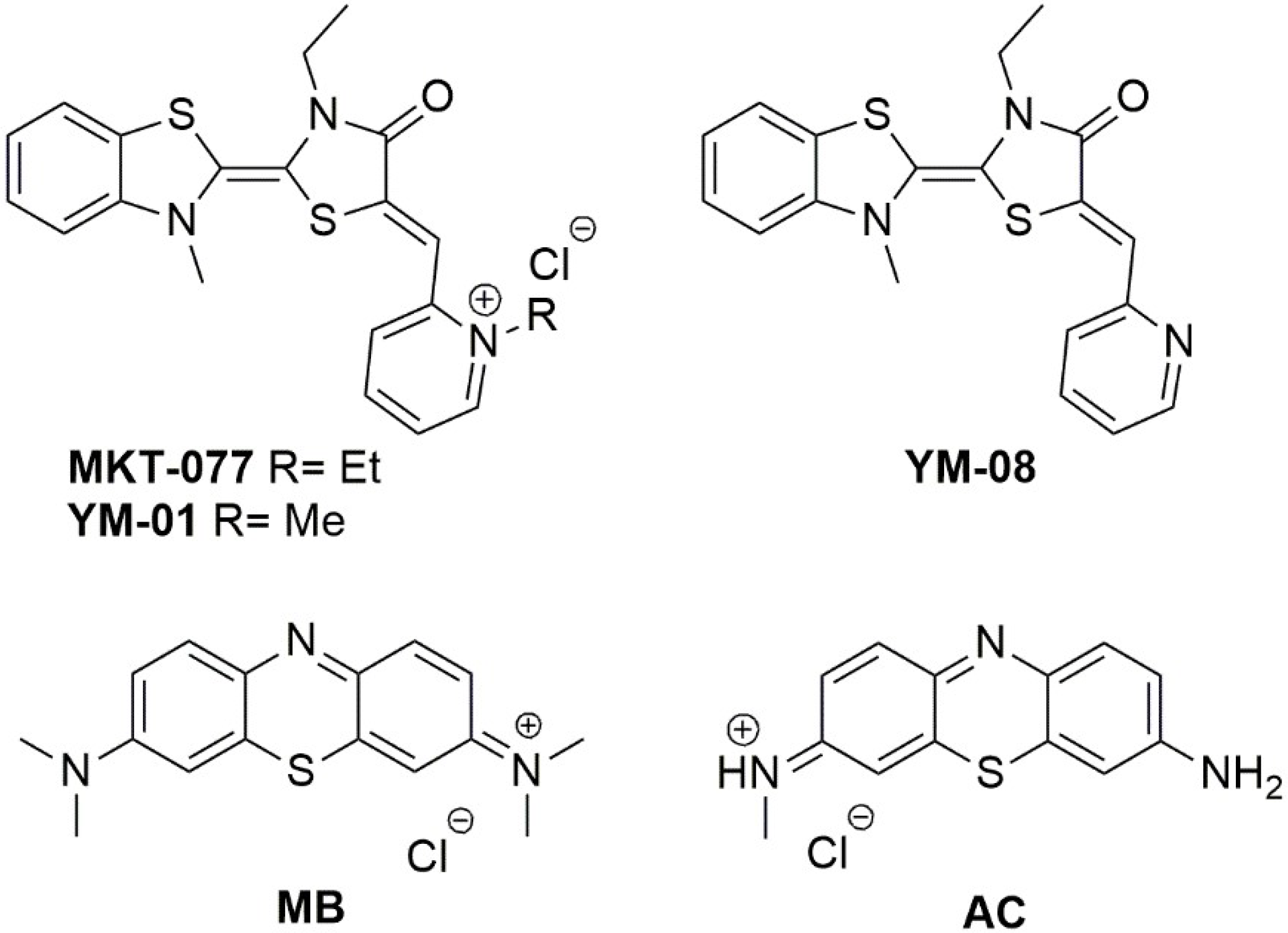
- Rousaki, A.; Miyata, Y.; Jinwal, U.K.; Dickey, C.A.; Gestwicki, J.E.; Zuiderweg, E.R. Allosteric drugs: The interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J. Mol. Biol. 2011, 411, 614–632. [Google Scholar] [CrossRef] [PubMed]
- Abisambra, J.; Jinwal, U.K.; Miyata, Y.; Rogers, J.; Blair, L.; Li, X.; Seguin, S.P.; Wang, L.; Jin, Y.; Bacon, J.; et al. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol. Psychiatry 2013, 74, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.M.; Baker, J.D.; Suntharalingam, A.; Nordhues, B.A.; Shelton, L.B.; Zheng, D.; Sabbagh, J.J.; Haystead, T.A.J.; Gestwicki, J.E.; Dickey, C.A. Inhibition of Both Hsp70 Activity and Tau Aggregation in Vitro Best Predicts Tau Lowering Activity of Small Molecules. ACS Chem. Biol. 2016, 11, 2041–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, Y.; Li, X.; Lee, H.F.; Jinwal, U.K.; Srinivasan, S.R.; Seguin, S.P.; Young, Z.T.; Brodsky, J.L.; Dickey, C.A.; Sun, D.; et al. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem. Neurosci. 2013, 4, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Koren, J.; O’Leary, J.C.; Jones, J.R.; Abisambra, J.F.; Dickey, C.A. Hsp70 ATPase Modulators as Therapeutics for Alzheimer’s and other Neurodegenerative Diseases. Mol. Cell Pharmacol. 2010, 2, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Cascio, F.; Kayed, R.; Azure, C. Targets and Modulates Toxic Tau Oligomers. ACS Chem. Neurosci. 2018, 9, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
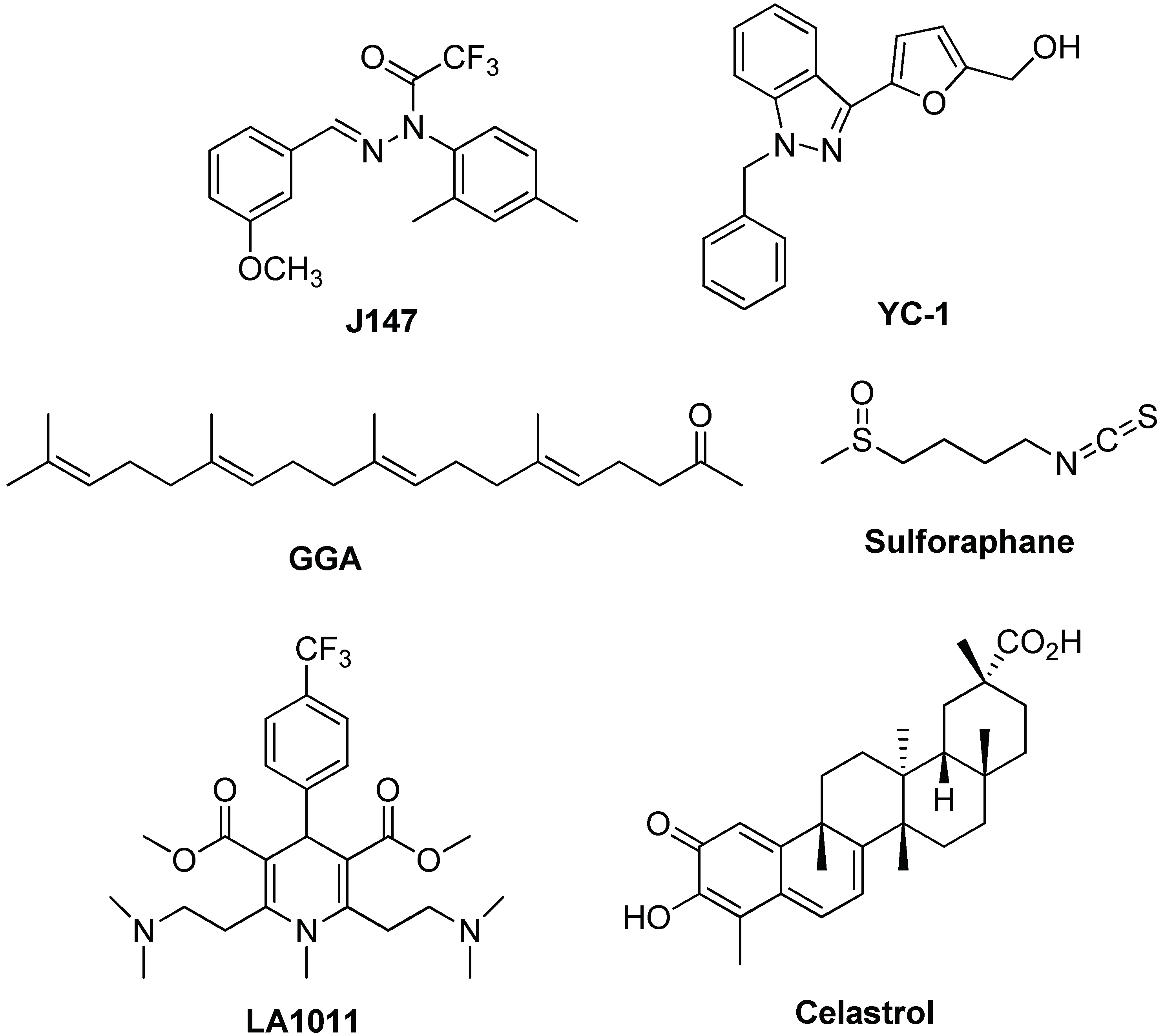
- Chen, Q.; Prior, M.; Dargusch, R.; Roberts, A.; Riek, R.; Eichmann, C.; Chiruta, C.; Akaishi, T.; Abe, K.; Maher, P.; et al. A Novel Neurotrophic Drug for Cognitive Enhancement and Alzheimer’s Disease. PLoS ONE 2011, 6, e27865. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Lee, Y.M.; Lam, K.K.; Lin, J.F.; Wang, J.J.; Yen, M.H.; Cheng, P.Y. The Role of Heat Shock Protein 70 in the Protective Effect of YC-1 on β-Amyloid-Induced Toxicity in Differentiated PC12 Cells. PLoS ONE 2013, 8, e69320. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Suzuki, K.; Matsushima, T.; Yamakawa, N.; Suzuki, T.; Mizushima, T. Suppression of Alzheimer’s Disease-Related Phenotypes by Geranylgeranylacetone in Mice. PLoS ONE 2013, 8, e76306. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Choi, B.R.; Kim, J.; LaFerla, F.M.; Yoon Park, J.H.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane upregulates the heat shock protein co-chaperone CHIP and clears amyloid-β and tau in a mouse model of Alzheimer’s disease. Mol. Nutr. Food. Res. 2018, 62, 1800240. [Google Scholar] [CrossRef] [PubMed]
- Kasza, Á.; Hunya, Á.; Frank, Z.; Fülőp, F.; Tőrők, Z.; Balogh, G.; Sántha, M.; Bálind, A.; Bernáth, S.; Blundell, L.I.M.K.; et al. Dihydropyridine Derivatives Modulate Heat Shock Responses and have a Neuroprotective Effect in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2016, 53, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, C.; Kalionis, B.; Wan, W.; Murthi, P.; Chen, C.; Li, Y.; Xia, S. EGb761 protects against Aβ1−42 oligomer-induced cell damage via endoplasmic reticulum stress activation andHsp70 protein expression increase in SH-SY5Y cells. Exp. Gerontol. 2016, 75, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.A.; Tang, D.W.F.; Hanif, A.; Brown, I.R. Induction of heat shock proteins in cerebral cortical cultures by celastrol. Cell Stress Chaperones 2013, 18, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.R.; Tan, M.S.; Xie, A.M.; Yu, J.T.; Tan, L. Heat shock protein 90 in Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 796869. [Google Scholar] [CrossRef] [PubMed]
- Kakimura, J.; Kitamura, Y.; Takata, K.; Umeki, M.; Suzuki, S.; Shibagaki, K.; Taniguchi, T.; Nomura, Y.; Gebicke-Haerter, P.J.; Smith, M.A.; et al. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 2002, 16, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Liu, T.; Zhao, X.; Trotter, C.; Yrigoin, K.; Cazzaro, S.; Narvaez, E.; Khan, H.; Witas, R.; Bukhari, A.; et al. Enhanced tau pathology via RanBP9 and Hsp90/Hsc70 chaperone complexes. Hum. Mol. Genet. 2017, 26, 3973–3988. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, Q.; Alam, M.Z.; Sait, K.H.W.; Anfinan, N.; Noorwali, A.W.; Kamal, M.A.; Khan, M.S.A.; Haque, A. Translational Shift of HSP90 as a Novel Therapeutic Target from Cancer to Neurodegenerative Disorders: An Emerging Trend in the Cure of Alzheimer’s and Parkinson’s Diseases. Curr. Drug Metabol. 2017, 18, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Biamonte, M.A.; Van de Water, R.; Arndt, J.W.; Scannevin, R.H.; Perret, D.; Lee, W.C. Heat shock protein 90: Inhibitors in clinical trials. J. Med. Chem. 2010, 14, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Shax, E.; Walter, J.G.; Märzhäuser, H.; Sthal, F.; Scheper, T.; Agard, D.A.; Eichner, S.; Kirschning, A.; Zeilinger, C. Microarray-based screening of heat shock protein inhibitors. J. Biotechnol. 2014, 180, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lauria, A.; Abbate, I.; Gentile, C.; Angileri, F.; Martorana, A.; Almerico, A.M. Synthesis and biological activities of a new class of heat shock protein 90 inhibitors, designed by energy-based pharmacophore virtual screening. J. Med. Chem. 2013, 56, 3424–3428. [Google Scholar] [CrossRef] [PubMed]
- Shumaila, K.; Subhankar, P. Identifying a C-terminal ATP-binding sites-based novel Hsp90-Inhibitor in silico: A plausible therapeutic approach in Alzheimer’s disease. Med. Hypotheses 2014, 83, 39–46. [Google Scholar] [CrossRef]
- Dickey, C.A.; Eriksen, J.; Kamal, A.; Burrows, F.; Kasibhatla, S.; Eckman, C.B.; Hutton, M.; Petrucelli, L. Development of a High Throughput Drug Screening Assay for the Detection of Changes in Tau Levels—Proof of Concept with HSP90 inhibitors. Curr. Alzheimer Res. 2005, 2, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Ortega, L.; Calvillo, M.; Luna, F.; Pérez-Severiano, F.; Rubio-Osornio, M.; Guevara, G.; Limon, I.D. 17-AAG improves cognitive process and increases heat shock protein response in a model lesion with Aβ25-35. Neuropeptides 2014, 48, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.W.; Tsui, Y.T.C.; Wong, T.T.; Cheung, S.K.K.; Goggins, W.B.; Yi, L.M.; Cheng, K.K.; Baum, L. Effect of 17-allylamino-17-demethoxygeldanamy-cin(17-AAG) in transgenic mouse models of frontotemporal lobar degeneration and Alzheimer’s disease. Transl. Neurodegener. 2013, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, B.; Liu, D.; Li, J.J.; Xue, Y.; Sakata, K.; Zhu, L.Q.; Heldt, S.A.; Xu, H.; Liao, F.F. Hsp90 Chaperone Inhibitor 17-AAG Attenuates Aβ-Induced Synaptic Toxicity and Memory Impairment. J. Neurosci. 2014, 34, 2464–2470. [Google Scholar] [CrossRef] [PubMed]
- Sinadinos, C.; Quraishe, S.; Sealey, M.; Samson, P.B.; Mudher, A.; Wyttenbach, A. Low Endogenous and Chemical Induced Heat Shock Protein Induction in a 0N3Rtau-Expressing Drosophila Larval Model of Alzheimer’s Disease. J. Alzheimers Dis. 2013, 33, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Pillay, I.; Nakano, H.; Sharma, S.V. Radicicol inhibits tyrosine phosphorylation of the mitotic Src substrate Sam68 and retards subsequent exit from mitosis of Src-transformed cells. Cell Growth Differ. 1996, 7, 1487–1499. [Google Scholar] [PubMed]
- Lu, Y.; Ansar, S.; Michaelis, M.L.; Blagg, B.S.J. Neuroprotective activity and evaluation of Hsp inhibitors in an immortalized neuronal cell line. Bioorg. Med. Chem. 2009, 17, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Ansar, S.; Burlison, J.A.; Kyle Hadden, M.; Ming Yu, X.; Desino, K.E.; Bean, J.; Neckers, L.; Audus, K.L.; Michaelis, M.L.; Blagg, B.S.J. A non-toxic Hsp90 inhibitor protects neurons from Aβ-induced toxicity. Bioorg. Med. Chem. Lett. 2007, 17, 1984–1990. [Google Scholar] [CrossRef] [PubMed]
- Thirstrup, K.; Sotty, F.; Pereira Montezinho, L.C.; Badolo, L.; Thougaard, A.; Kristjansson, M.; Jensen, T.; Watson, S.; Nielsen, S.M. Linking HSP90 target occupancy to HSP70 induction and efficacy in mouse brain. Pharmacol. Res. 2016, 104, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Y.; Huang, L.; Chen, J.; Li, J.J.; Wang, R.; Kim, E.; Chen, Y.; Justicia, C.; Sakata, K.; et al. A CNS-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer’s mouse model via an HSF1-mediated mechanism. Mol. Psychiatry 2017, 22, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Shelton, B.L.; Baker, J.D.; Zheng, D.; Sullivan, L.E.; Solanki, P.K.; Webster, J.M.; Sun, Z.; Sabbagh, J.J.; Nordhues, B.A.; Koren, J., III; et al. Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc. Natl. Acad. Sci. USA 2017, 144, 9707–9712. [Google Scholar] [CrossRef] [PubMed]
- Narayan, M.; Zhang, J.; Braswell, K.; Gibson, C.; Zitnyar, A.; Lee, D.C.; Varghese-Gupta, S.; Jinwal, U.K. Withaferin A regulates LRRK2 levels by interfering with the Hsp90-Cdc37 chaperone complex. Curr. Aging Sci. 2015, 8, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Kim, J.H.; Chang, B.J.; Zweckstetter, M. Mechanistic basis for the recognition of a misfolded protein by the molecular chaperone Hsp90. Nat. Stuct. Mol. Biol. 2017, 24, 407–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radli, M.; Rüdiger, S.G.D. Dancing with the Diva: Hsp90- Client interactions. J. Mol. Biol. in press. [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diseases | Proteins Involved | Reference |
|---|---|---|
| AD | Aβ and Tau | [20] |
| PD | α-Synuclein and Tau | [3] |
| Huntington | Huntingtin | [33] |
| Prion | PrP | [34] |
| Taupathies | Tau | [3] |
| Lewy bodies dementia | α-Synuclein and ubiquitin | [35] |
| HSPs | Localization | Association | Functions | CNS Diseases | Pharmacological targeting |
|---|---|---|---|---|---|
| Hsp60 |
| - APP/Aβ [41,66,67]. |
| ||
| Hsp70 |
|
| - AD [31,98,99,100]. |
| |
| Hsp90 |
| - Aβ [43]. | - AD [43]. |
|
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campanella, C.; Pace, A.; Caruso Bavisotto, C.; Marzullo, P.; Marino Gammazza, A.; Buscemi, S.; Palumbo Piccionello, A. Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting. Int. J. Mol. Sci. 2018, 19, 2603. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092603
Campanella C, Pace A, Caruso Bavisotto C, Marzullo P, Marino Gammazza A, Buscemi S, Palumbo Piccionello A. Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting. International Journal of Molecular Sciences. 2018; 19(9):2603. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092603
Chicago/Turabian StyleCampanella, Claudia, Andrea Pace, Celeste Caruso Bavisotto, Paola Marzullo, Antonella Marino Gammazza, Silvestre Buscemi, and Antonio Palumbo Piccionello. 2018. "Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting" International Journal of Molecular Sciences 19, no. 9: 2603. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092603