DNA Methylation Analysis of Dormancy Release in Almond (Prunus dulcis) Flower Buds Using Epi-Genotyping by Sequencing


 , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Results
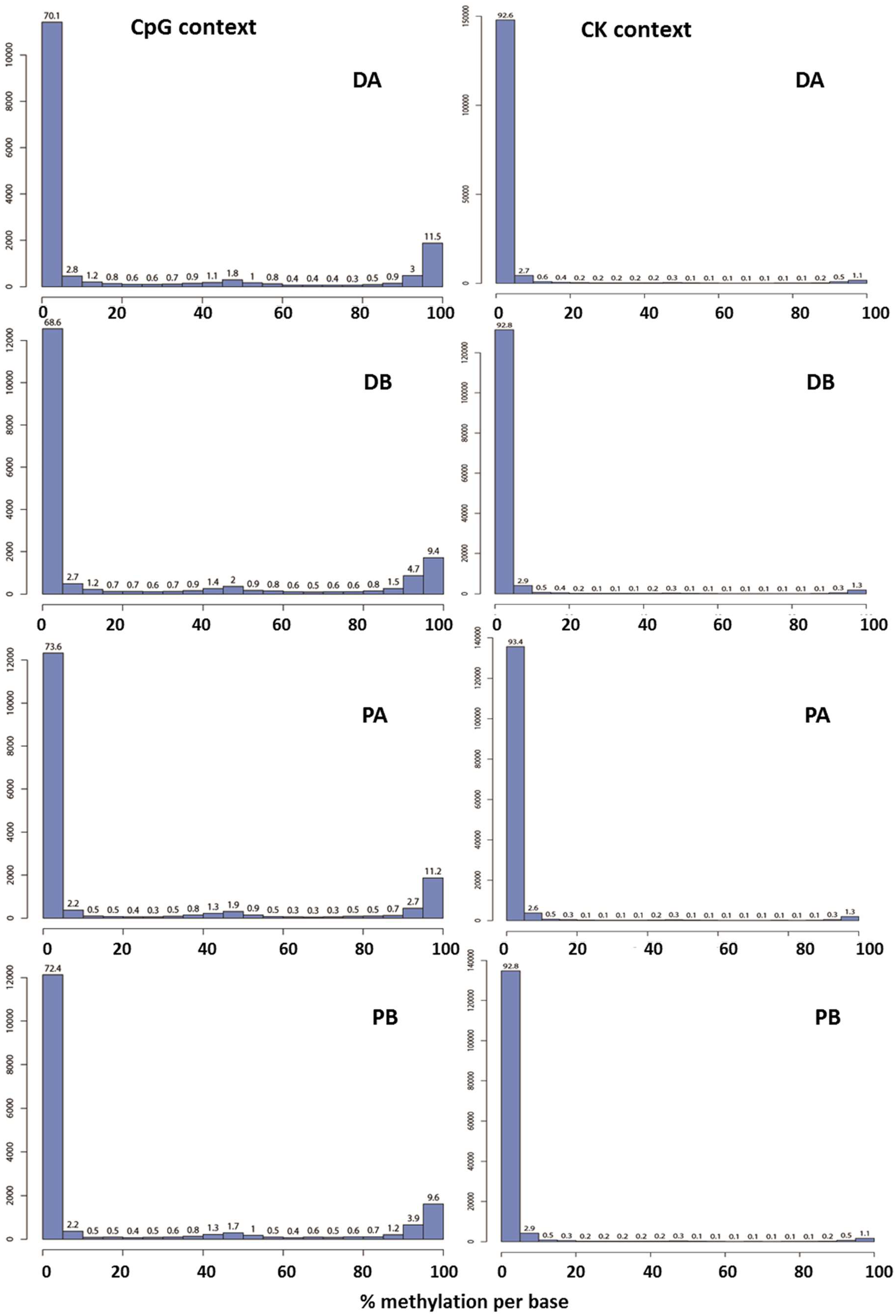
2.1. Evaluation of the Quality of the Epi-GBS Analysis
2.2. Differentyally Methylated Genes Detected
2.3. Differentyally Methylated Genes Related to Bud Dormancy
3. Discussion
4. Materials and Methods
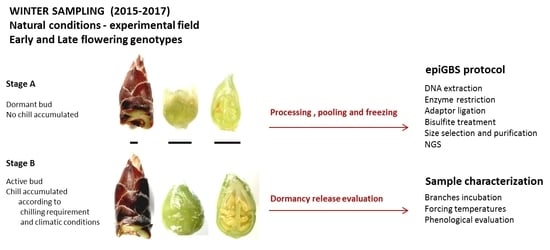
4.1. Plant Material and Experimental Design
4.2. Epi-GBS Protocol
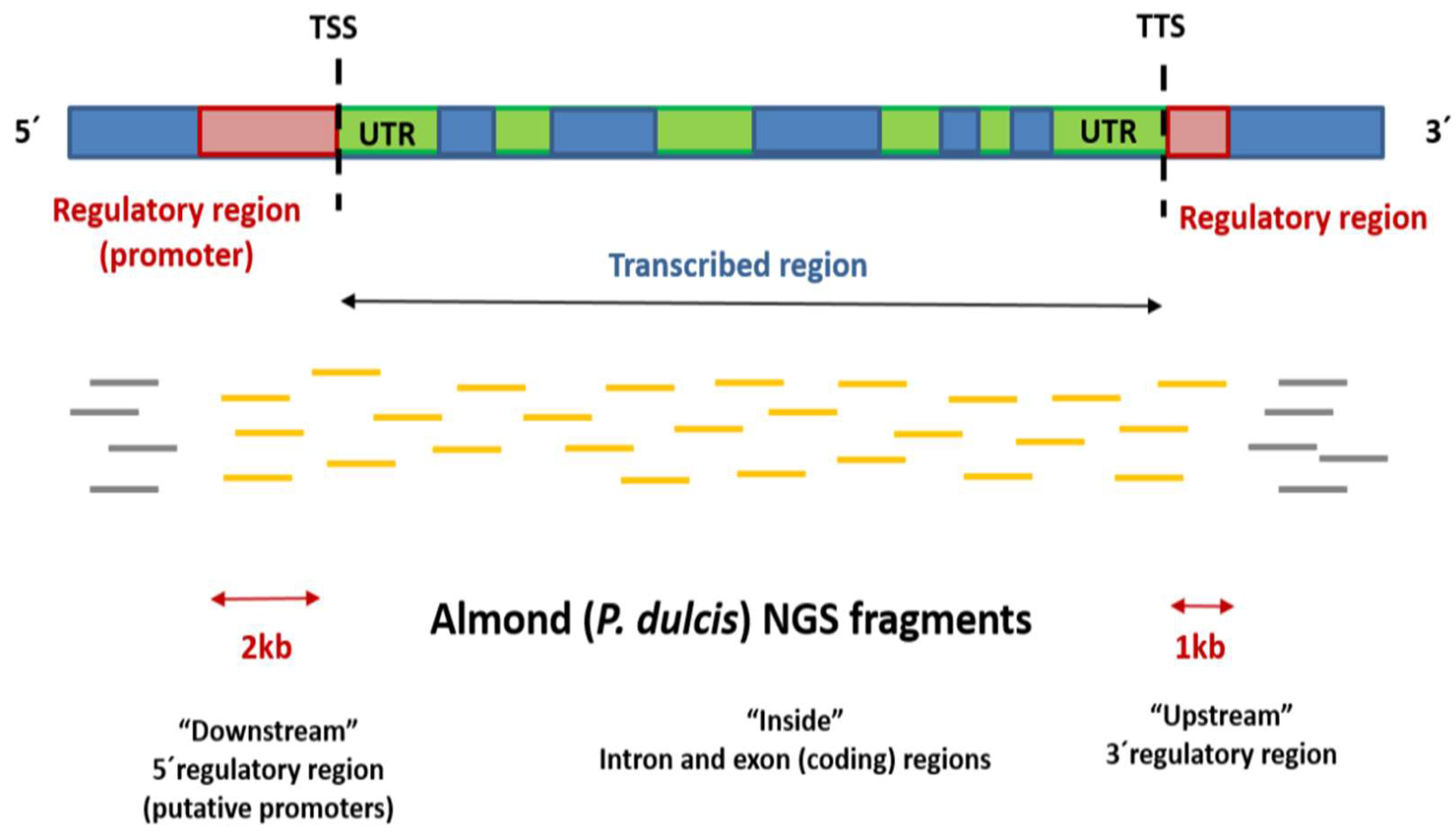
4.3. Bioinformatic Analysis of DNA Methylation
4.4. Gene Finding and Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5mC | 5′ Methylated Cytosine |
| AP180 | ASSEMBLY PROTEIN 180 |
| ARF | AUXIN RESPONSE FACTOR |
| CytP450 | Cytochrome P-450 |
| DMF | Differentially Methylated Fragment |
| DMG | Differentially Methylated Gene |
| epiGBS | Epi-Genotyping By Sequencing |
| FAR1 | FAR-RED IMPAIRED RESPONSE 1 |
| GDSL | Gly, Asp, Ser, and Leu motif |
| GO | Gene Ontology |
| GPI | Glycerophosphatidylinositol |
| H3K27me3 | Trymethylation of Histone 3 on Lis (K) residue 27 |
| HSP | HYDROPHOBIC SEED PROTEIN |
| LEA | LATE EMBRYOGENESIS ABUNDANT |
| LHY | LATE ELONGATED HYPOCOTYL |
| LRR-TIR | LEUCINE RICH REPEAT-TOLL INTERLEUKIN 1 RECEPTOR |
| MADS | MCM1-AGAMOUS-DEFICIENS-SRF |
| MAPK | MITOGEN-ACTIVATED PROTEIN KINASE |
| NGS | Next Generation Sequencing |
| NF-Y | NUCLEAR FACTOR-Y |
| PTMs | Post-Translational Modifications |
| SAM | S-ADENOSYL METHIONINE |
| VPS1 | VACUOLAR PROTEIN SORTING 1 |
| WDSAM1 | Tryptophan-Aspartic acid-Sterile Alpha Motif 1 |
References
- Vitasse, Y.; Lenz, A.; Körner, C. The interaction between freezing tolerance and phenology in temperate deciduous trees. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauvieux, R.; Wenden, B.; Dirlewanger, E. Bud Dormancy in Perennial Fruit Tree Species: A Pivotal Role for Oxidative Cues. Front. Plant Sci. 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.A.; Early, J.D.; Martin, G.C.; Darnell, R.L. Endo-, para-, and eco-dormancy physiological terminology and classification for dormancy research. HortScience 1987, 22, 371–377. [Google Scholar]
- Egea, J.; Ortega, E.; Martínez-Gómez, P.; Dicenta, F. Chilling and heat requirements of almond cultivars for flowering. Environ. Exp. Bot. 2003, 50, 79–85. [Google Scholar] [CrossRef]
- Martínez-Gómez, P.; Prudencio, A.S.; Gradziel, T.M.; Dicenta, F. The delay of flowering time in almond: A review of the combined effect of adaptation, mutation and breeding. Euphytica 2017, 213, 197. [Google Scholar] [CrossRef]
- Prudencio, A.S.; Martínez-Gómez, P.; Dicenta, F. Evaluation of breaking dormancy, flowering and productivity of extra-late and ultra-late flowering almond cultivars during cold and warm seasons in South-East of Spain. Sci. Hort. 2018, 235, 39–46. [Google Scholar] [CrossRef]
- Cooke, J.E.K.; Eriksson, M.E.; Juntilla, O. The dynamic nature of bud dormancy in trees: Environmental control and molecular mechanisms. Plant Cell Environ. 2012, 35, 1707–1728. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.G.; Zhebentyayeva, T.; Barakat, A.; Liu, Z. The genetic control of bud-break in trees. Adv. Bot. Res. 2015, 74, 201–228. [Google Scholar]
- Yaish, M.W.; Colasanti, J.; Rothstein, S.J. The role of epigenetics processes in controlling flowering time exposed to stress. J. Exp. Bot. 2011, 62, 3727–3735. [Google Scholar] [CrossRef] [PubMed]
- Ríos, G.; Leida, C.; Conejero, C.; Badenes, M.L. Epigenetic regulation of bud dormancy events in perennial plants. Front. Plant Sci. 2014, 5, 247. [Google Scholar] [PubMed]
- Saze, H. Epigenetic memory transmission through mitosis and meiosis in plants. Semin. Cell Dev. Biol. 2008, 19, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Jacobsen, S.E. Epigenetic modifications in plants: An evolutionary perspective. Curr. Opin. Plant Biol. 2011, 14, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Cañal, M.J.; Correia, B.; Escandon, M.; Hasbún, R.; Meijón, M.; Pinto, G.; Valledor, L. Can Epigenetics Help Forest Plants to Adapt to Climate Change? In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications: Transcriptional Regulation and Chromatin Remodelling in Plants; Alvarez-Venegas, R., De la Peña, C., Casas-Mollano, J.A., Eds.; Springer: Cham, Switzerland, 2014; pp. 125–146. [Google Scholar]
- Santamaría, M.; Hasbún, R.; Valera, M.; Meijón, M.; Valledor, L.; Rodríguez, J.L.; Rodríguez, R. Acetylated H4 histone and genomic DNA methylation patterns during bud set and bud burst in Castanea sativa. J. Plant Physiol. 2009, 166, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, L.; Conesa, A.; Lloret, A.; Badenes, M.L.; Ríos, G. Genome-wide changes in histone H3 lysine 27 trimethylation associated with bud dormancy release in peach. Tree Genet. Gen. 2015, 11, 45. [Google Scholar] [CrossRef]
- Lloret, A.; Martínez-Fuentes, A.; Agustí, M.; Badenes, M.L.; Ríos, G. Chromatin-associated regulation of sorbitol synthesis in flower buds of peach. Plant Mol. Biol. 2017, 95, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Rothkegel, K.; Sánchez, E.; Montes, C.; Greve, M.; Tapia, S.; Bravo, S.; Almeida, A.M. DNA methylation and small interference RNAs participate in the regulation of MADS-box genes involved in dormancy in sweet cherry (Prunus avium L.). Tree Physiol. 2017, 37, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Rattan, U.K.; Singh, A.K. Chilling-Mediated DNA Methylation Changes during Dormancy and Its Release Reveal the Importance of Epigenetic Regulation during Winter Dormancy in Apple (Malus × domestica Borkh.). PLoS ONE 2016, 11, e0149934. [Google Scholar] [CrossRef] [PubMed]
- Prudencio, A.S.; Dicenta, F.; Martínez-Gómez, P. Gene expression analysis of flower bud dormancy breaking in almond using RNA-Seq. In Proceedings of the VII International Symposium on Almonds & Pistachios, Adelaida, Australia, 5–9 November 2017. [Google Scholar]
- Fernández i Martí, A.; Gradziel, T.M.; Socias i Company, R. Methylation of the Sf locus in almond is associated with S-RNase loss of function. Plant Mol. Biol. 2014, 86, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Fresnedo-Ramírez, J.; Chan, H.M.; Parfitt, D.E.; Crisosto, C.H.; Gradziel, T.M. Genome-wide DNA-(de) methylation is associated with Noninfectious Bud-failure exhibition in Almond (Prunus dulcis [Mill.] D.A.Webb). Sci. Rep. 2017, 7, 42686. [Google Scholar]
- van Gurp, T.P.; Wagemaker, N.V.; Wouters, B.; Vergeer, P.; Ouborg, J.N.; Werhoeben, K.J.V. epiGBS: Reference-free reduced representation bisulfite sequencing. Nat. Meth. 2016, 13, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L. Widespread natural variation of DNA methylation within angiosperms. Gen. Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- How-Kit, A.; Emeline, T.; Deleuze, J.F.; Gallusci, P. Locus-Specific DNA Methylation Analysis and Applications to Plants. In Plant Epigenetics; Rajewsky, N., Jurga, S., Barciszewski, J., Eds.; Springer: Berlin, Germany, 2017; pp. 303–328. [Google Scholar]
- Kawakatsu, T.; Huang, S.S.; Jupe, F. Epigenomic diversity in a global collection of Arabidopsis thaliana accesions. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Vining, K.J.; Pomraning, K.R.; Wilhelm, L.J.; Priest, H.D.; Pellegrini, M.; Mockler, T.C. Dynamic DNA cytosine methylation in the Populus trichocarpa genome: Tissue-level variation and relationship to gene expression. BMC Gen. 2012, 13, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Custard, K.D.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell 2006, 18, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Montgomery, T.A.; Fahlgren, N.; Kasschau, K.D.; Nonogaki, H.; Carrington, J.C. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 2007, 52, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-Z.; Jin, S.-H.; Li, P.; Jiang, X.-Y.; Li, Y.-J.; Hou, B.K. Ectopic expression of UGT84A2 delayed flowering by indole-3-butyric acid-mediated transcriptional repression of ARF6 and ARF8 genes in Arabidopsis. Plant Cell Rep. 2017, 36, 1995–2006. [Google Scholar] [CrossRef] [PubMed]
- Bañuelos, M.L.G.; Moreno, L.V.; Winzerling, J.; Orozco, J.A.; Gardea, A.A. Winter metabolism in deciduous trees: Mechanisms, genes and associated proteins. Rev. Fitotec. Mex. 2008, 31, 295–308. [Google Scholar]
- Du, D.; Zhang, Q.; Cheng, T.; Pan, H.; Yang, W.; Sun, L. Genome-wide identification and analysis of late embryogenesis abundant (LEA) genes in Prunus mume. Mol. Biol. Rep. 2013, 40, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Yamane, H.; Kashiwa, Y.; Kakehi, E.; Yonemori, K.; Mori, H.; Hayashi, K.; Iwamoto, K.; Tao, R.; Kataoka, I. Differential expression of dehydrin in flower buds of two Japanese apricot cultivars requiring different chilling requirements for bud break. Tree Physiol. 2006, 26, 1559–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, S.; Oda, A.; Yoshida, R.; Niinuma, K.; Miyata, K.; Tomozoe, Y.; Tajima, T.; Nakagawa, M.; Hayashi, K.; Coupland, G.; et al. Circadian Clock Proteins LHY and CCA1 Regulate SVP Protein Accumulation to Control Flowering in Arabidopsis. Plant Cell 2008, 20, 2960–2971. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Eo, H.J.; Jang, I.-C.; Kang, H.-G.; Song, J.T.; Seo, H.S. Ubiquitination of LHY by SINAT5 regulates flowering time and is inhibited by DET1. Biochem. Biophys. Res. Commun. 2010, 398, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-J.; Kwon, Y.-J.; Gil, K.-E.; Park, C.-M. LATE ELONGATED HYPOCOTYL regulates photoperiodic flowering via the circadian clock in Arabidopsis. BMC Plant Biol. 2016, 16, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varriale, A. DNA Methylation in Plants and Its Implications in development, Hybrid Vigour, and Evolution. In Plant Epigenetics; Rajewsky, N., Jurga, S., Barciszewski, J., Eds.; Springer: Berlin, Germany, 2017; pp. 263–280. [Google Scholar]
- Viggiano, L.; de Pinto, M.C. Dynamic DNA Methylation Patterns in Stress Response. In Plant Epigenetics; Rajewsky, N., Jurga, S., Barciszewski, J., Eds.; Springer: Berlin, Germany, 2017; pp. 281–302. [Google Scholar]
- Garg, R.; Narayana Chevala, V.V.S.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 2015, 5, 14922. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Pourkheirandish, M.; Morishige, H.; Kubo, Y.; Nakamura, M.; Ichimura, K.; Seo, S.; Kanamori, H.; Wu, J.; Ando, T.; et al. Mitogen-Activated Protein Kinase Kinase 3 Regulates Seed Dormancy in Barley. Curr. Biol. 2016, 26, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Aravind, L. Origin and evolution of eukaryotic apoptosis: The bacterial connection. Cell Death Differ. 2002, 9, 394. [Google Scholar] [CrossRef] [PubMed]
- Del Duca, S.; Serafini-Fracassini, D.; Cai, G. Senescence and programmed cell death in plants: Polyamine action mediated by transglutaminase. Front. Plant Sci. 2014, 5, 120. [Google Scholar] [CrossRef] [PubMed]
- Ebine, K.; Ueda, T. Roles of membrane trafficking in plant cell wall dynamics. Front. Plant Sci. 2015, 6, 878. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Brandizzi, F. The plant secretory pathway: An essential factory for building the plant cell wall. Plant Cell Physiol. 2014, 55, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Rinne, P.L.H.; Welling, A.; Vahala, J.; Ripel, L.; Ruonala, R.; Kangasjärvi, J.; van der Schoot, C. Chilling of Dormant Buds Hyperinduces FLOWERING LOCUS T and Recruits GA-Inducible 1,3-β-Glucanases to Reopen Signal Conduits and Release Dormancy in Populus. Plant Cell 2011, 23, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Felipe, A.J. Phenological states of almond. In Proceedings of the Third GREMPA Colloquium, Bari, Italy, 3–7 October 1977; pp. 101–103. (In Italian). [Google Scholar]
- Doyle, J.J.; Doyle, M. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Verde, I.; Jenkins, J.; Dondini, L.; Micali, S.; Pagliarani, G.; Vendramin, E. The Peach v2.0 release: High-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Gen. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Yue, L.; Hengyu, Y.; Qi, Y.; Xin, Y.; Zhou, D.; Wenying, X.; Zhen, S. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Differentially Methylated Fragments | ||
|---|---|---|
| D–P genotype comparison | ||
| Hypo (<) | Hyper (>) | Stages |
| 307 | 370 | A and B |
| 677 DMFs | ||
| A–B stage comparison | ||
| Hypo (<) | Hyper (>) | Genotype |
| 3 | 20 | D |
| 21 | 27 | P |
| 3 | 7 | D and P |
| 10 DMFs | ||
| Differentially Methylated Genes | |||
|---|---|---|---|
| Methylation State | Gene Position | Gene Hits | Genes Identified |
| Hypermethylated | Upstream | 36 | |
| Inside | 291 | ||
| Downstream | 134 | ||
| Total | 461 | 423 | |
| Hypomethylated | Upstream | 19 | |
| Inside | 201 | ||
| Downstream | 80 | ||
| Total | 300 | 281 | |
| Equally-methylated | Upstream | 6 | |
| Inside | 41 | ||
| Downstream | 8 | ||
| Total | 55 | 27 | |
| Total DMGs | 816 | 731 | |
| Methylation State | Gene Position | Differentially Methylated Genes | ||
|---|---|---|---|---|
| ‘Desmayo Largueta’ | ‘Penta‘ | Common | ||
| Hyper-methylated | Upstream | 7 | 3 | 1 |
| Inside | 5 | 2 | 2 | |
| Downstream | 5 | 9 | 4 | |
| Total | 17 | 14 | 7 | |
| Hypo-methylated | Upstream | 1 | - | - |
| Total | 18 | 14 | 7 | |
| FragmentID | Comparison | Chromosome | Gene Position | Prupe.Gene Code |
|---|---|---|---|---|
| fragment1289 | AB | Pp01 | Downstream | Prupe.1G287200 |
| fragment1294 | AB | Pp01 | Upstream | Prupe.1G125600 |
| fragment3735 | PAPB | Pp01 | Downstream | Prupe.1G099900 |
| fragment32 | AB | Pp03 | Downstream | Prupe.3G130700 |
| fragment341 | AB | Pp06 | Downstream | Prupe.6G307900 |
| fragment797 | AB | Pp02 | Inside | Prupe.2G074400 |
| fragment1708 | DADB | Pp02 | Inside | Prupe.2G019300 |
| fragment4206 | PAPB | Pp02 | Inside | Prupe.2G019300 |
| fragment341 | DADB | Pp02 | Downstream | Prupe.2G031100 |
| fragment255 | DADB | Pp02 | Upstream | Prupe.2G053300 |
| fragment92 | DADB | Pp02 | Inside | Prupe.2G057100 |
| fragment92 | DADB | Pp02 | Inside | Prupe.2G057800 |
| fragment797 | DADB | Pp02 | Upstream | Prupe.2G074300 |
| fragment3707 | PAPB | Pp04 | Downstream | Prupe.4G186400 |
| fragment4154 | PAPB | Pp04 | Downstream | Prupe.4G253800 |
| fragment238 | AB | Pp04 | Downstream | Prupe.4G270800 |
| fragment3299 | PAPB | Pp06 | Downstream | Prupe.6G307900 |
| fragment1289 | DADB | Pp02 | Inside | Prupe.2G146000 |
| fragment341 | DADB | Pp03 | Upstream | Prupe.3G026400 |
| fragment797 | DADB | Pp03 | Downstream | Prupe.3G130700 |
| fragment2263 | DADB | Pp05 | Inside | Prupe.5G036900 |
| fragment157 | PAPB | Pp06 | Upstream | Prupe.6G014500 |
| fragment1289 | DADB | Pp05 | Downstream | Prupe.5G038500 |
| fragment483 | PAPB | Pp02 | Upstream | Prupe.2G039500 |
| fragment507 | DADB | Pp06 | Inside | Prupe.6G097800 |
| fragment1708 | DADB | Pp06 | Upstream | Prupe.6G331300 |
| fragment2727 | DADB | Pp06 | Upstream | Prupe.6G331300 |
| fragment849 | PAPB | Pp01 | Downstream | Prupe.1G105700 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prudencio, Á.S.; Werner, O.; Martínez-García, P.J.; Dicenta, F.; Ros, R.M.; Martínez-Gómez, P. DNA Methylation Analysis of Dormancy Release in Almond (Prunus dulcis) Flower Buds Using Epi-Genotyping by Sequencing. Int. J. Mol. Sci. 2018, 19, 3542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113542
Prudencio ÁS, Werner O, Martínez-García PJ, Dicenta F, Ros RM, Martínez-Gómez P. DNA Methylation Analysis of Dormancy Release in Almond (Prunus dulcis) Flower Buds Using Epi-Genotyping by Sequencing. International Journal of Molecular Sciences. 2018; 19(11):3542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113542
Chicago/Turabian StylePrudencio, Ángela S., Olaf Werner, Pedro J. Martínez-García, Federico Dicenta, Rosa M. Ros, and Pedro Martínez-Gómez. 2018. "DNA Methylation Analysis of Dormancy Release in Almond (Prunus dulcis) Flower Buds Using Epi-Genotyping by Sequencing" International Journal of Molecular Sciences 19, no. 11: 3542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113542