Niacin in the Central Nervous System: An Update of Biological Aspects and Clinical Applications


Abstract
:1. Introduction
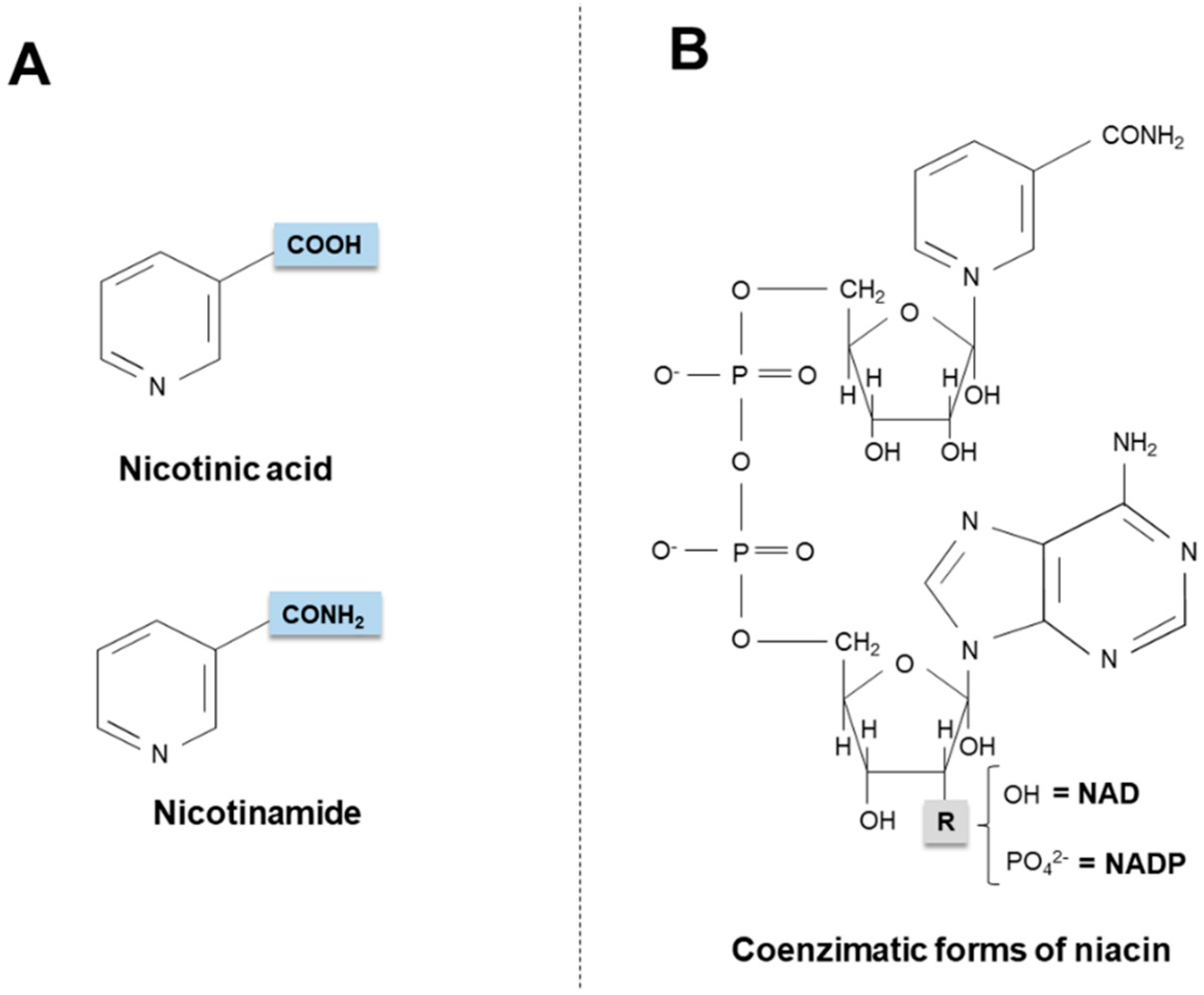
2. Niacin Sources
2.1. Exogenous Sources
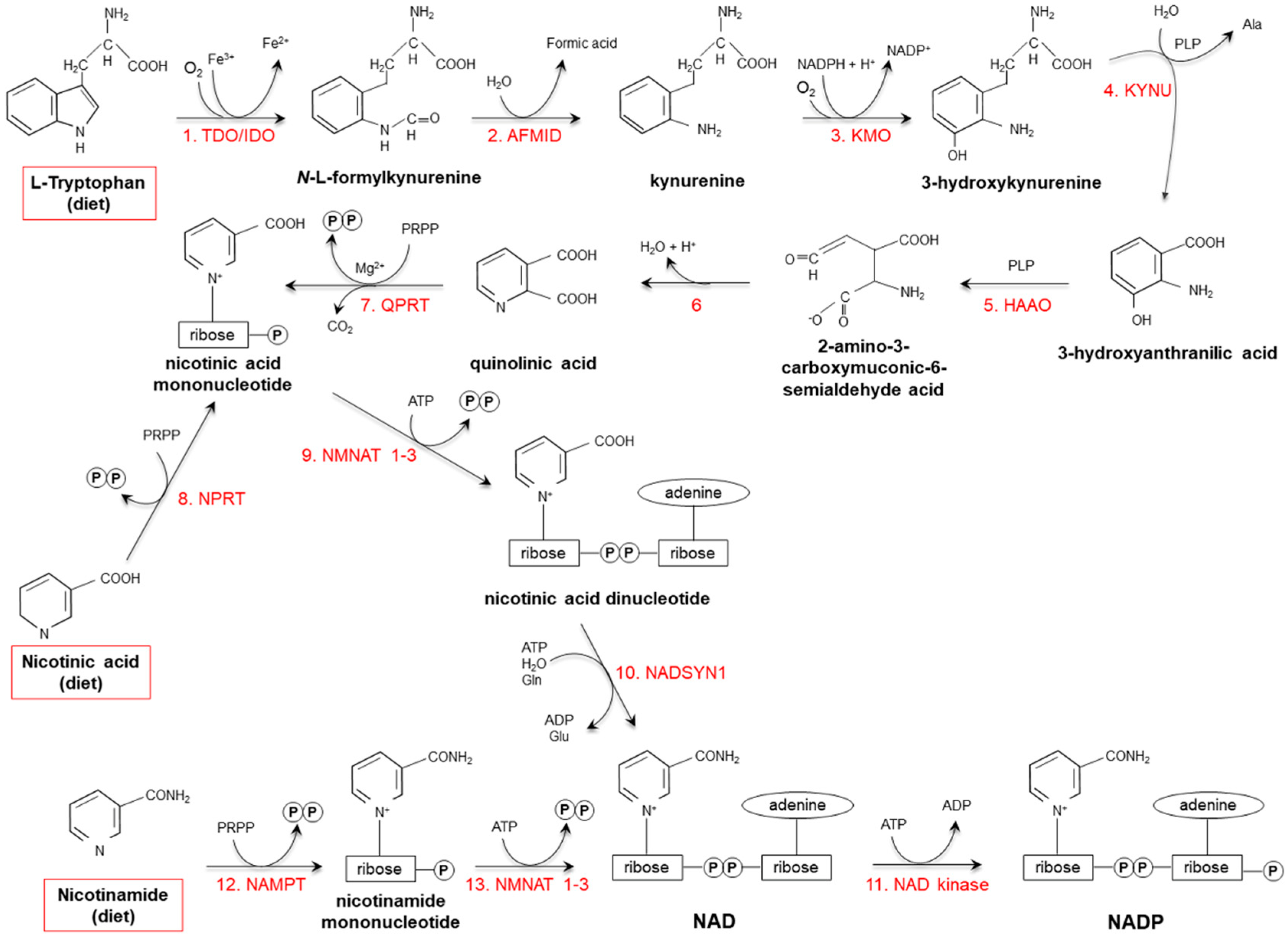
2.2. Endogenous Synthesis
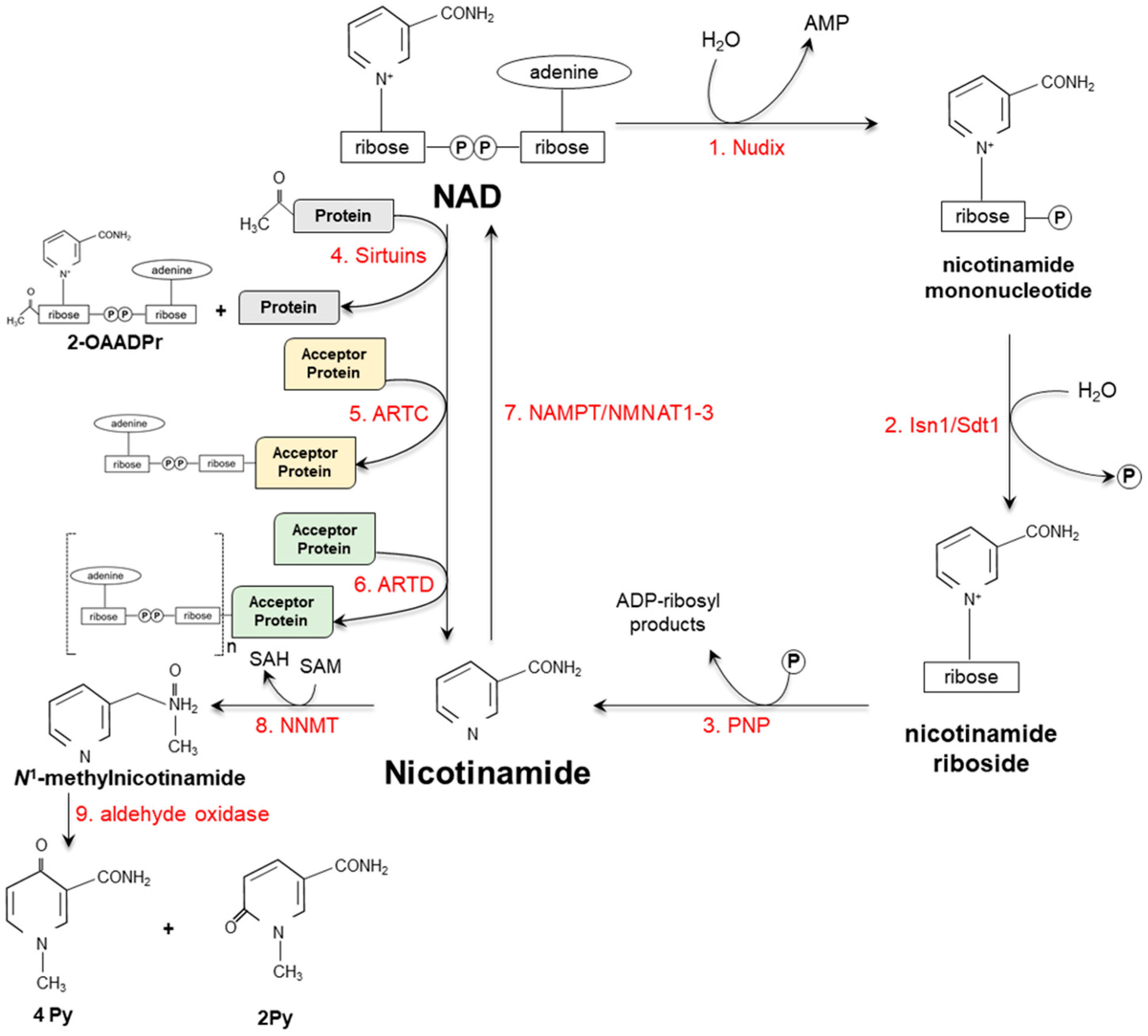
3. Vitamin Catabolism
4. Severe Vitamin Deficiency
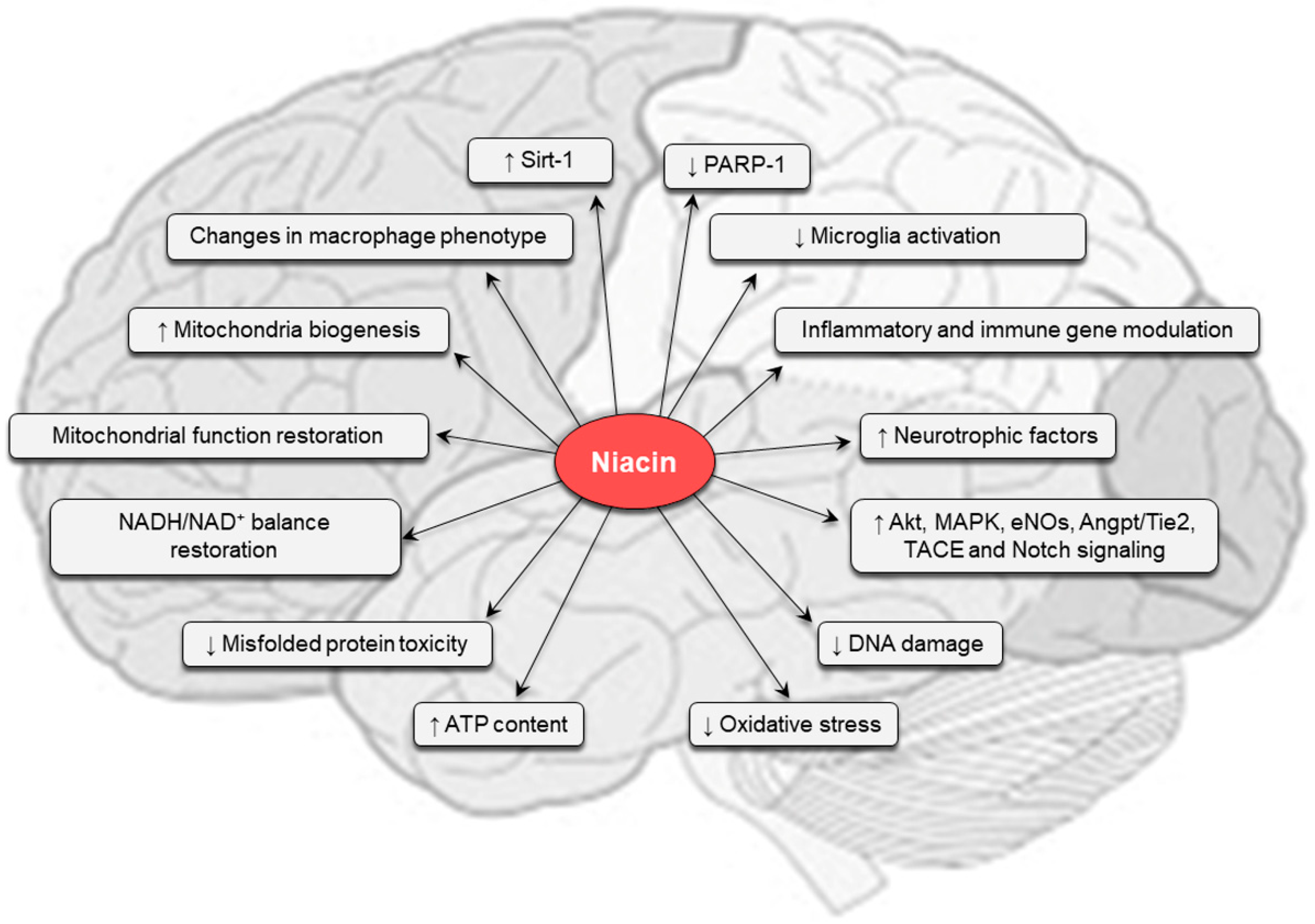
5. Pharmacological Effects of Niacin
6. Niacin in the Central Nervous System
7. Alzheimer’s Disease
8. Parkinson’s Disease
9. Huntington’s Disease
10. Other Neurological Diseases
10.1. Ischemic and Traumatic Injuries
10.2. Headache
10.3. Psychiatric Disorders
11. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Akt | protein kinase B |
| ARTC | ADP-ribosyltransferases |
| ARTD | diphtheria toxin-like ADP-ribosyltransferases |
| CNS | central nervous system |
| FOXO3a | forkhead transcription factor |
| HD | Huntington’s disease |
| HDL | high density lipoprotein |
| hOAT-10 | human organic anion transporter-10 |
| Htt | huntingtin |
| IDO | indolamine-pyrrole 2-3 dioxygenase |
| KP | kynurenine pathway |
| LDL | low density lipoprotein |
| MDD | major depressive disorder |
| MPP+ | N-methy-l-4-phenylpyridinium |
| NAD(P) | nicotinamide adenine dinucleotide (phosphate) |
| NAMPT | nicotinamide phosphoribosyltransferase |
| NE | niacin equivalents |
| NMDA | N-methyl-D-aspartate |
| NNMT | N-methyltransferase |
| PARP | poly(ADP-ribose) polymerase |
| PD | Parkinson’s disease |
| polyQ | polyglutamine repeat |
| ROS | reactive oxygen species |
| SAM | S-adenosyl-methionine |
| SIRT | sirtuin |
| SMCT1/SLC5A8 | sodium-coupled monocarboxylate transporter |
| TBI | traumatic brain injury |
| TDO | tryptophan 2,3 dioxygenase |
| Trp | tryptophan |
| VLDL | very low density lipoprotein |
References
- Spies, T.D.; Bean, W.B.; Stone, R.E. The Treatment of Subclinical and Classic Pellagra Use of Nicotinic Acid, Nicotinic Acid Amide and Sodium Nicotinate, with Special Reference to the Vasodilator Action and the Effect on Mental Symptoms. JAMA 1938, 111, 584–592. [Google Scholar] [CrossRef]
- Magni, G.; Amici, A.; Emanuelli, M.; Orsomando, G.; Raffaelli, N.; Ruggieri, S. Enzymology of NAD+ Homeostasis in Man. Cell. Mol. Life Sci. 2004, 61, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.P.; Calvo, S.E.; Mootha, V.K. Spatiotemporal Compartmentalization of Hepatic NADH and NADPH Metabolism. J. Biol. Chem. 2018, 293, 7508–7516. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.B. Niacin Status, NAD Distribution and ADP-Ribose Metabolism. Curr. Pharm. Des. 2009, 15, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, A.; Kulikova, V.; Ziegler, M. The Human NAD Metabolome: Functions, Metabolism and Compartmentalization. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Madsen, A.S.; Olsen, C.A.; Hirschey, M.D. Metabolic Control by Sirtuins and Other Enzymes that Sense NAD(+), NADH, or Their Ratio. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Mutafova-Yambolieva, V.N.; Hwang, S.J.; Hao, X.; Chen, H.; Zhu, M.X.; Wood, J.D.; Ward, S.M.; Sanders, K.M. Beta-Nicotinamide Adenine Dinucleotide is an Inhibitory Neurotransmitter in Visceral Smooth Muscle. Proc. Natl. Acad. Sci. USA 2007, 104, 16359–16364. [Google Scholar] [CrossRef] [PubMed]
- Moreschi, I.; Bruzzone, S.; Nicholas, R.A.; Fruscione, F.; Sturla, L.; Benvenuto, F.; Usai, C.; Meis, S.; Kassack, M.U.; Zocchi, E.; De Flora, A. Extracellular NAD+ Is an Agonist of the Human P2Y11 Purinergic Receptor in Human Granulocytes. J. Biol. Chem. 2008, 281, 31419–31429. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Grahnert, A.; Abdelrahman, A.; Muller, C.E.; Hauschildt, S. Extracellular NAD(+) Induces a Rise in [Ca(2+)](i) in Activated Human Monocytes via Engagement of P2Y(1) and P2Y(11) Receptors. Cell Calcium 2009, 46, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Stone, T.W. The Kynurenine Pathway and the Brain: Challenges, Controversies and Promises. Neuropharmacology 2016, 112, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Prousky, J.; Millman, C.; Kirkland, J. Pharmacologic Use of Niacin. J. Evid.-Based Complement. Altern. Med. 2011, 16, 91–101. [Google Scholar] [CrossRef]
- Bahn, A.; Hagos, Y.; Reuter, S.; Balen, D.; Brzica, H.; Krick, W.; Burckhardt, B.C.; Sabolic, I.; Burckhardt, G. Identification of a New Urate and High Affinity Nicotinate Transporter, hOAT10 (SLC22A13). J. Biol. Chem. 2008, 283, 16332–16341. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.S.; Subramanian, V.S.; Kapadia, R.; Kashyap, M.L.; Said, H.M. Mammalian Colonocytes Possess a Carrier-Mediated Mechanism for Uptake of Vitamin B3 (niacin): Studies Utilizing Human and Mouse Colonic Preparations. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Gopal, E.; Miyauchi, S.; Martin, P.M.; Ananth, S.; Roon, P.; Smith, S.B.; Ganapathy, V. Transport of Nicotinate and Structurally Related Compounds by Human SMCT1 (SLC5A8) and Its Relevance to Drug Transport in the Mammalian Intestinal Tract. Pharm. Res. 2007, 24, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Gopal, E.; Fei, Y.J.; Miyauchi, S.; Zhuang, L.; Prasad, P.D.; Ganapathy, V. Sodium-Coupled and Electrogenic Transport of B-Complex Vitamin Nicotinic Acid by slc5a8, a Member of the Na/glucose Co-transporter Gene Family. Biochem. J. 2005, 388, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Spector, R. Niacinamide Transport Through the Blood-Brain Barrier. Neurochem. Res. 1987, 12, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A. Tryptophan Metabolism in Alcoholism. Nutr. Res. Rev. 2002, 15, 123–152. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Müller, F. Flavin-Dependent Hydroxylases. Biochem. Soc. Trans. 1985, 13, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Kobayashi, R.; Fukuwatari, T. Vitamin B1 Deficiency Inhibits the Increased Conversion of Tryptophan to Nicotinamide in Severe Food-Restricted Rats. Biosci. Biotechnol. Biochem. 2015, 79, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Onodera, M. Comparison of Tryptophan-Niacin Conversion in Rats Fed with a Nicotinic Acid-Free Diet Containing Egg White, Egg White Proteolysate, or Mixtures of Amino Acids. Agric. Biol. Chem. 1991, 55, 1291–1298. [Google Scholar] [CrossRef]
- Shibata, K.; Nomamoto, R.; Iwai, K. Effect of Dietary Protein levels on the Urinary Excretion of Nicotinamide and Its Metabolites in Rats. Agric. Biol. Chem. 1988, 53, 1765–1769. [Google Scholar] [CrossRef]
- Shibata, K. Nutritional Factors that Regulate on the Conversion of L-Tryptophan to Niacin. Adv. Exp. Med. Biol. 1999, 467, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K. Organ Co-Relationship in Tryptophan Metabolism and Factors That Govern the Biosynthesis of Nicotinamide from Tryptophan. J. Nutr. Sci. Vitaminol. 2018, 64, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Nakata, C.; Fukuwatari, T. Moderate Food Restriction Suppresses the Conversion of L-tryptophan to Nicotinamide in Weaning Rats. Biosci. Biotechnol. Biochem. 2014, 78, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A. Pellagra and Alcoholism: A Biochemical Perspective. Alcohol Alcohol. 2014, 49, 238–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, D.A. Inhibition in Vitro of the Enzymes of the Oxidative Pathway of Tryptophan Metabolism and of Nicotinamide Nucleotide Synthesis by Benserazide, Carbidopa and Isoniazid. Biochem. Pharmacol. 1980, 29, 707–712. [Google Scholar] [CrossRef]
- Braidman, I.P.; Rose, D.P. The Effect of Sex Hormones on the Activity of Tryptophan Oxygenase and Other Corticosteroid-Inducible Enzymes in Rat Liver. Biochem. J. 1971, 122, 28P. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Shinno, H.; Ichihara, A. Insulin and Glucagon as a New Regulator System for Tryptophan Oxygenase Activity Demonstrated in Primary Cultured Rat Hepatocytes. J. Biol. Chem. 1980, 255, 7533–7535. [Google Scholar] [PubMed]
- Nakamura, T.; Niimi, S.; Nawa, K.; Noda, C.; Ichihara, A.; Takagi, Y.; Anai, M.; Sakaki, Y. Multihormonal Regulation of Transcription of the Tryptophan 2,3-Dioxygenase Gene in Primary Cultures of Adult Rat Hepatocytes with Special Reference to the Presence of a Transcriptional Protein Mediating the Action of Glucocorticoids. J. Biol. Chem. 1987, 262, 723–732. [Google Scholar]
- Labrie, F.; Korner, A. Effect of Glucagon, Insulin, and Thyroxine on Tyrosine Transaminase and Tryptophan Pyrrolase of Rat Liver. Arch. Biochem. Biophys. 1969, 129, 75–78. [Google Scholar] [CrossRef]
- Chiancone, F.M. Enzyme Activities in the Tryptophan → Nicotinic Acid Path in Physiopathology. Ital. J. Biochem. 1964, 13, 1–30. [Google Scholar]
- Ku, Y.; Rogers, Q.R.; Harper, A.E. Effects of Thyroxine and Cortisol on Liver Threonine Dehydratase and Tryptophan Pyrrolase in Rats Fed a High Protein Diet. Proc. Soc. Exp. Biol. Med. 1969, 130, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Toda, S. Effect of Thyroxine on the Metabolism of Tryptophan to Nicotinamide in Rats. Biosci. Biotechnol. Biochem. 1994, 58, 1757–1762. [Google Scholar] [CrossRef]
- Horwitt, M.K.; Harper, A.E.; Henderson, L.M. Niacin-Tryptophan Relationships for Evaluating Niacin Equivalents. Am. J. Clin. Nutr. 1981, 34, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Food and Nutrition Board. Dietary Reference Intakes for Thiamine, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Panthotenic Acid, Biotin and Choline; National Academy Press: Washington, DC, USA, 1998; pp. 374–389. [Google Scholar]
- Long, A.; Klimova, N.; Kristian, T. Mitochondrial NUDIX hydrolases: A Metabolic Link Between NAD Catabolism, GTP and Mitochondrial Dynamics. Neurochem. Int. 2017, 109, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Bogan, K.L.; Evans, C.; Belenky, P.; Song, P.; Burant, C.F.; Kennedy, R.; Brenner, C. Identification of Isn1 and Sdt1 as Glucose- and Vitamin-Regulated Nicotinamide Mononucleotide and Nicotinic Acid Mononucleotide [corrected] 5′-Nucleotidases Responsible for Production of Nicotinamide Riboside and Nicotinic Acid Riboside. J. Biol. Chem. 2009, 284, 34861–34869. [Google Scholar] [CrossRef]
- Wielgus-Kutrowska, B.; Kulikowska, E.; Wierzchowski, J.; Bzowska, A.; Shugar, D. Nicotinamide Riboside, an Unusual, Non-Typical, Substrate of Purified Purine-Nucleoside Phosphorylases. Eur. J. Biochem. 1997, 243, 408–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 Inhibits Glutamate Dehydrogenase and Opposes the Effects of Calorie Restriction in Pancreatic Beta Cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 Promotes DNA Repair Under Stress by Activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; He, J.; Liao, M.; Hu, M.; Li, W.; Ouyang, H.; Wang, X.; Ye, T.; Zhang, Y.; Ouyang, L. An Overview of Sirtuins as Potential Therapeutic Target: Structure, Function and Modulators. Eur. J. Med. Chem. 2019, 161, 48–77. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. Ten Years of NAD-Dependent SIR2 Family Deacetylases: Implications for Metabolic Diseases. Trends Pharmacol. Sci. 2010, 31, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-Pinzon, M.A. Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front. Endocrinol. 2018, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Chang, P. Insights Into the Biogenesis, Function, and Regulation of ADP-Ribosylation. Nat. Chem. Biol. 2018, 14, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Kunze, F.A.; Hottiger, M.O. Regulating Immunity via ADP-Ribosylation: Therapeutic Implications and Beyond. Trends Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Crawford, K.; Bonfiglio, J.J.; Mikoč, A.; Matic, I.; Ahel, I. Specificity of Reversible ADP-Ribosylation and Regulation of Cellular Processes. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural Determination of a Cyclic Metabolite of NAD+ with Intracellular Ca2+-Mobilizing Activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar] [PubMed]
- Swarbrick, J.M.; Graeff, R.; Garnham, C.; Thomas, M.P.; Galione, A.; Potter, B.V. ‘Click Cyclic ADP-Ribose’: A Neutral Second Messenger Mimic. Chem. Commun. 2014, 50, 2458–2461. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, E.; Lo Buono, N.; Horenstein, A.L.; Funaro, A.; Malavasi, F. The ADP-Ribosyl Cyclases--the Current Evolutionary State of the ARCs. Front. Biosci. 2014, 19, 986–1002. [Google Scholar] [CrossRef]
- Wei, W.; Graeff, R.; Yue, J. Roles and Mechanisms of the CD38/Cyclic Adenosine Diphosphate Ribose/Ca(2+) Signaling Pathway. World J. Biol. Chem. 2014, 5, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Harlan, B.A.; Killoy, K.M.; Vargas, M.R. Nicotinamide Adenine Dinucleotide Metabolism and Neurodegeneration. Antioxid. Redox Signal. 2018, 28, 1652–1668. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, J.P.; Bowlby, S.C.; Wheeler, F.B.; Shi, L.; Sui, G.; Davis, A.L.; Howard, T.D.; D’Agostino, R.B., Jr.; Miller, L.D.; Sirintrapun, S.J.; et al. CD38 Inhibits Prostate Cancer Metabolism and Proliferation by Reducing Cellular NAD(+) Pools. Mol. Cancer Res. 2018, 11, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Horenstein, A.L.; Rizzo, R.; Malavasi, F. The Role of Extracellular Adenosine Generation in the Development of Autoimmune Diseases. Mediators Inflamm. 2018, 2018, 7019398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ying, W. NAD(+) Deficiency Is a Common Central Pathological Factor of a Number of Diseases and Aging: Mechanisms and Therapeutic Implications. Antioxid. Redox Signal. 2018, 30, 890–905. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. NNMT Activation Can Contribute to the Development of Fatty Liver Disease by Modulating the NAD (+) Metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.H.; Kim, D.; Bae, S.Y.; Kim, W.K.; Hong, J.Y.; Lee, H.J.; Rajasekaran, N.; Kwon, S.; Fan, Y.; Luu, T.T.; et al. Targeting Nicotinamide N-Methyltransferase and miR-449a in EGFR-TKI-Resistant Non-Small-Cell Lung Cancer Cells. Mol. Ther. Nucleic Acids 2018, 11, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Crujeiras, A.B.; Pissios, P.; Moreno-Navarrete, J.M.; Diaz-Lagares, A.; Sandoval, J.; Gomez, A.; Ricart, W.; Esteller, M.; Casanueva, F.F.; Fernandez-Real, J.M. An Epigenetic Signature in Adipose Tissue Is Linked to Nicotinamide N-Methyltransferase Gene Expression. Mol. Nutr. Food Res. 2018, 62, e1700933. [Google Scholar] [CrossRef] [PubMed]
- Kannt, A.; Rajagopal, S.; Kadnur, S.V.; Suresh, J.; Bhamidipati, R.K.; Swaminathan, S.; Hallur, M.S.; Kristam, R.; Elvert, R.; Czech, J.; et al. A Small Molecule Inhibitor of Nicotinamide N-Methyltransferase for the Treatment of Metabolic Disorders. Sci. Rep. 2018, 8, 3660. [Google Scholar] [CrossRef] [PubMed]
- Rudolphi, B.; Zapp, B.; Kraus, N.A.; Ehebauer, F.; Kraus, B.J.; Kraus, D. Body Weight Predicts Nicotinamide N-Methyltransferase Activity in Mouse Fat. Endocr. Res. 2018, 43, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Nejabati, H.R.; Mihanfar, A.; Pezeshkian, M.; Fattahi, A.; Latifi, Z.; Safaie, N.; Valiloo, M.; Jodati, A.R.; Nouri, M. N1-Methylnicotinamide (MNAM) as a Guardian of Cardiovascular System. J. Cell. Physiol. 2018, 233, 6386–6394. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, K.; Parker, J.A. Nicotinamide-N-Methyltransferase Controls Behavior, Neurodegeneration and Lifespan by Regulating Neuronal Autophagy. PLoS Genet. 2018, 14, e1007561. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.M.; Long, H. Nicotinamide N-Methyltransferase as a Potential Marker for Cancer. Neoplasma 2018, 65, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.I.; Gömpel, K.; Bakuradze, T.; Eisenbrand, G.; Richling, E. Urinary Excretion of Niacin Metabolites in Humans After Coffee Consumption. Mol. Nutr. Food Res. 2018, 62, 1700735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Zhang, D.; Wang, X.; Zhang, L.; Han, J.; Yang, M.; Xiao, X.; Zhang, Y.; Liu, H. Simultaneous Quantification of Niacin and Its Three Main Metabolites in Human Plasma by LC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 904, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Shi, G.; Sui, Y.; Li, Q.; Tang, Y.; Gu, J. Quantification of Niacin and Its Metabolite Nicotinuric Acid in Human Plasma by LC-MS/MS: Application to a Clinical Trial of a Fixed Dose Combination Tablet of Niacin Extended-Release/Simvastatin (500 mg/10 mg) in Healthy Chinese Volunteers. Int. J. Anal. Chem. 2015, 2015, 212437. [Google Scholar] [CrossRef] [PubMed]
- Pittelli, M.; Formentini, L.; Faraco, G.; Lapucci, A.; Rapizzi, E.; Cialdai, F.; Romano, G.; Moneti, G.; Moroni, F.; Chiarugi, A. Inhibition of Nicotinamide Phosphoribosyltransferase: Cellular Bioenergetics Reveals a Mitochondrial Insensitive NAD Pool. J. Biol. Chem. 2010, 285, 34106–34114. [Google Scholar] [CrossRef] [PubMed]
- Sydenstricker, V.P. The History of Pellagra, Its Recognition as a Disorder of Nutrition and Its Conquest. Am. J. Clin. Nutr. 1958, 6, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Gentilcore, D. Louis Sambon and the Clash of Pellagra Etiologies in Italy and the United States, 1905–1914. J. Hist. Med. Allied Sci. 2016, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.B.; Zempleni, J.; Suttie, J.W.; Gregory, J.F., III; Stover, P.J. (Eds.) Handbook of Vitamins, 5th ed.; CRC Press: Boca Raton, FL, USA, 2013; pp. 149–190. [Google Scholar]
- Jagielska, G.; Tomaszewicz-Libudzic, E.C.; Brzozowska, A. Pellagra: A Rare Complication of Anorexia Nervosa. Eur. Child. Adolesc. Psychiatry 2007, 16, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, J.P.; da Cunha, D.F.; Filho, D.C.; Silva-Vergara, M.L.; dos Santos, V.M.; da Costa, J.C., Jr.; Etchebehere, R.M.; Gonçalves, J.; de Carvalho da Cunha, S.F.; Jordão, A.A.; et al. Niacin Metabolite Excretion in Alcoholic Pellagra and AIDS Patients with and without Diarrhea. Nutrition 2004, 20, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Li, T.; Wu, S.; Li, W.Q.; Weinstock, M.; Qureshi, A.A.; Cho, E. Niacin Intake and Risk of Skin Cancer in US Women and Men. Int. J. Cancer 2017, 140, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.B. Niacin Status and Treatment-Related Leukemogenesis. Mol. Cancer Ther. 2009, 8, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Rosmaninho, A.; Sanches, M.; Fernandes, I.C.; Pinto-Almeida, T.; Vilaça, S.; Oliveira, A.; Selores, M. Letter: Pellagra as the Initial Presentation of Crohn Disease. Dermatol. Online J. 2012, 18, 12. [Google Scholar] [PubMed]
- Prakash, R.; Gandotra, S.; Singh, L.K.; Das, B.; Lakra, A. Rapid Resolution of Delusional Parasitosis in Pellagra with Niacin Augmentation Therapy. Gen. Hosp. Psychiatry 2008, 30, 581–584. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization; United Nations High Commissions for Refugees. Pellagra and Its Prevention and Control in Major Emergencies. World Health Organization. 2000. Available online: http://www.who.int/nutrition/publications/emergencies/WHO_NHD_00.10/en/ (accessed on 1 December 2018 ).
- Altschul, R.; Hoffer, A.; Stephen, J.D. Influence of Nicotinic Acid on Serum Cholesterol in Man. Arch. Biochem. Biophys. 1955, 54, 558–559. [Google Scholar] [CrossRef]
- Zeman, M.; Vecka, M.; Perlík, F.; Staňková, B.; Hromádka, R.; Tvrzická, E.; Širc, J.; Hrib, J.; Žák, A. Pleiotropic Effects of Niacin: Current Possibilities for Its Clinical Use. Acta Pharm. 2016, 66, 449–469. [Google Scholar] [CrossRef] [PubMed]
- la Paz, S.M.; Bermudez, B.; Naranjo, M.C.; Lopez, S.; Abia, R.; Muriana, F.J. Pharmacological Effects of Niacin on Acute Hyperlipemia. Curr. Med. Chem. 2016, 23, 2826–2835. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S.; Colletti, S.L.; Lovenberg, T.W.; Semple, G.; Wise, A.; IJzerman, A.P. International Union of Basic and Clinical Pharmacology. LXXXII: Nomenclature and Classification of Hydroxy-carboxylic Acid Receptors (GPR81, GPR109A, andGPR109B). Pharmacol. Rev. 2011, 63, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Felts, A.S. Molecule of the Month. TREDAPTIVE (Nicotinic Acid/Laropiprant): A New Lipid-Modifying Therapy for the Treatment of LDL-C, HDL-C and Triglycerides. Curr. Top. Med. Chem. 2008, 8, 1310. [Google Scholar] [CrossRef] [PubMed]
- AIM-HIGH Investigators; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in Patients with Low HDL Cholesterol Levels Receiving Intensive Statin Therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- HPS2-THRIVE Collaborative Group. HPS2-THRIVE Randomized Placebo-Controlled Trial in 25 673 High-Risk Patients of ER Niacin/Laropiprant: Trial Design, Pre-Specified Muscle and Liver Outcomes, and Reasons for Stopping Studytreatment. Eur. Heart J. 2013, 34, 1279–1291. [Google Scholar] [CrossRef]
- HPS2-THRIVE Collaborative Group; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of Extended-Release Niacin with Laropiprant in High-Risk Patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Graff, E.C.; Fang, H.; Wanders, D.; Judd, R.L. Anti-inflammatory Effects of the Hydroxycarboxylic Acid Receptor 2. Metabolism 2016, 65, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, X.; Wang, N.; Yin, G.; Ma, S.; Fu, Y.; Wei, C.; Chen, Y.; Xu, W. GPR109A Expression in the Murine Min6 Pancreatic Beta Cell Line, and Its Relation with Glucose Metabolism and Inflammation. Ann. Clin. Lab. Sci. 2015, 45, 315–322. [Google Scholar] [PubMed]
- Xu, X.; Lin, S.; Chen, Y.; Li, X.; Ma, S.; Fu, Y.; Wei, C.; Wang, C.; Xu, W. The Effect of Metformin on the Expression of GPR109A, NF-κB and IL-1β in Peripheral Blood Leukocytes from Patients with Type 2 Diabetes Mellitus. Ann. Clin. Lab. Sci. 2017, 47, 556–562. [Google Scholar] [PubMed]
- Heemskerk, M.M.; Dharuri, H.K.; van den Berg, S.A.; Jónasdóttir, H.S.; Kloos, D.P.; Giera, M.; van Dijk, K.W.; van Harmelen, V. Prolonged Niacin Treatment Leads to Increased Adipose Tissue PUFA Synthesis and Anti-Inflammatory Lipid and Oxylipin Plasmaprofile. J. Lipid Res. 2014, 55, 2532–2540. [Google Scholar] [CrossRef] [PubMed]
- Wanders, D.; Graff, E.C.; White, B.D.; Judd, R.L. Niacin Increases Adiponectin and Decreases Adipose Tissue Inflammation in High Fat Diet-Fed Mice. PLoS ONE 2013, 8, 71285. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Sun, G.; Liu, H.; Shu, L.; Zhang, J.; Guo, L.; Huang, C.; Xu, J. Niacin Suppresses Progression of Atherosclerosis by Inhibiting Vascular Inflammation and Apoptosis of Vascular Smooth Muscle Cells. Med. Sci. Monit. 2015, 21, 4081–4089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, E.; Li, Y.; Yao, M.; Wei, Z.; Fu, Y.; Yang, Z. Niacin Attenuates the Production of Pro-Inflammatory Cytokines in LPS-Induced Mouse Alveolar Macrophages by HCA2 Dependent Mechanisms. Int. Immunopharmacol. 2014, 23, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.; Kim, H.J.; Rodriguez-Iturbe, B.; Vaziri, N.D. Niacin ameliorates oxidative stress, inflammation, proteinuria, and hypertension in rats with chronic renal failure. Am. J. Physiol. Ren. Physiol. 2009, 297, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.Y.; Suh, G.J.; Kwon, W.Y.; Kim, K.S.; Jung, Y.S.; Kye, Y.C. The Therapeutic Effect and Mechanism of Niacin on Acute Lung Injury in a Rat Model of Hemorrhagic Shock: Down-Regulation of the Reactive Oxygen Species-Dependent Nuclear Factor κB Pathway. J. Trauma Acute Care Surg. 2015, 79, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Godin, A.M.; Ferreira, W.C.; Rocha, L.T.; Ferreira, R.G.; Paiva, A.L.; Merlo, L.A.; Nascimento, E.B., Jr.; Bastos, L.F.; Coelho, M.M. Nicotinic Acid Induces Antinociceptive and Anti-Inflammatory Effects in Different Experimental Models. Pharmacol. BiochemBehav. 2012, 101, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Ruddock, M.W.; Hirst, D.G. Nicotinamide Relaxes Vascular Smooth Muscle by Inhibiting Myosin Light Chain Kinase-Dependent Signaling Pathways: Implications for Anticancer Efficacy. Oncol. Res. 2004, 14, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Ruddock, M.W.; Burns, D.M.; McKeown, S.R.; Murphy, L.; Walsh, I.K.; Keane, P.F.; Hirst, D.G. Contractile Properties of Human Renal Cell Carcinoma Recruited Arteries and Theirresponse to Nicotinamide. Radiother. Oncol. 2000, 54, 179–184. [Google Scholar] [CrossRef]
- Agote, M.; Viaggi, M.; Kreimann, E.; Krawiec, L.; Dagrosa, M.A.; Juvenal, G.J.; Pisarev, M.A. Influence of Nicotinamide on the Radiosensitivity of Normal and Goitrous Thyroid in the Rat. Thyroid 2001, 11, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, P.; Rojas, A.; Saunders, M. Accelerated Radiotherapy, Carbogen, and Nicotinamide (ARCON) in the Treatment of Advanced Bladder Cancer: Mature Results of a Phase II Nonrandomized Study. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Ramsden, D. Nicotinamide: A Double Edged Sword. Parkinsonism Relat. Disord. 2005, 11, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Fricker, R.A.; Green, E.L.; Jenkins, S.I.; Griffin, S.M. The Influence of Nicotinamide on Health and Disease in the Central Nervous System. Int. J. Tryptophan Res. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Turski, W.A.; Nakamura, M.; Todd, W.P.; Carpenter, B.K.; Whetsell, W.O., Jr.; Schwarcz, R. Identification and Quantification of Kynurenic Acid in Human Brain Tissue. Brain Res. 1988, 454, 164–169. [Google Scholar] [CrossRef]
- Gobaille, S.; Kemmel, V.; Brumaru, D.; Dugave, C.; Aunis, D.; Maitre, M. Xanthurenic Acid Distribution, Transport, Accumulation and Release in the Rat Brain. J. Neurochem. 2008, 105, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Schwarcz, R. Presence of 3-hydroxyanthranilic Acid in Rat Tissues and Evidence for Its Production from Anthranilic Acid in the Brain. J. Neurochem. 1990, 55, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-brain Barrier Transport of Kynurenines: Implications for Brain Synthesis and Metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.C.; Collins, J.F.; Schwarcz, R. On the Excitotoxic Properties of Quinolinic Acid, 2,3-Piperidine Dicarboxylic Acids and Structurally Related Compounds. Neuropharmacology 1983, 22, 1331–1342. [Google Scholar] [CrossRef]
- Moroni, F.; Lombardi, G.; Carlà, V.; Moneti, G. The Excitotoxin Quinolinic Acid is Present and Unevenly Distributed in the Rat Brain. Brain Res. 1984, 19, 352–355. [Google Scholar] [CrossRef]
- Bohár, Z.; Toldi, J.; Fülöp, F.; Vécsei, L. Changing the Face of Kynurenines and Neurotoxicity: Therapeutic Considerations. Int. J. Mol. Sci. 2015, 16, 9772–9793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majewski, M.; Kozlowska, A.; Thoene, M.; Lepiarczyk, E.; Grzegorzewski, W.J. Overview of the Role of Vitamins and Minerals on the Kynurenine Pathway in Health and Disease. J. Physiol. Pharmacol. 2016, 67, 3–19. [Google Scholar] [PubMed]
- Dang, Y.; Dale, W.E.; Brown, O.R. Comparative Effects of Oxygen on Indoleamine 2,3-Dioxygenase and Tryptophan 2,3-Dioxygenase of the Kynurenine Pathway. Free Radic. Biol. Med. 2000, 28, 615–624. [Google Scholar] [CrossRef]
- Kanai, M.; Nakamura, T.; Funakoshi, H. Identification and Characterization of Novel Variants of the Tryptophan 2,3-Dioxygenase Gene: Differential Regulation in the Mouse Nervous System During Development. Neurosci. Res. 2009, 64, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An Endogenous Tumour-Promoting Ligand of the Human Aryl Hydrocarbon Receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Williams, S.K.; Stojic, A.; Iwantscheff, S.; Sonner, J.K.; Grabitz, C.; Becker, S.; Böhler, L.I.; Mohapatra, S.R.; Sahm, F.; et al. Tryptophan-2,3-Dioxygenase (TDO) Deficiency is Associated with Subclinical Neuroprotection in a Mouse Model of Multiplesclerosis. Sci. Rep. 2017, 7, 41271. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.C.; André, C.; Wang, Y.; Lawson, M.A.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Interferon-Gamma and Tumor Necrosis Factor-Alpha Mediate the Upregulation of Indoleamine 2,3-Dioxygenase and the Induction of Depressive-Like Behavior in Mice in Response to Bacillus Calmette-Guerin. J. Neurosci. 2009, 29, 4200–4209. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.C.; Lawson, M.A.; André, C.; Briley, E.M.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Herkenham, M.; Dantzer, R.; Kelley, K.W. Induction of IDO by Bacille Calmette-Guérin is Responsible for Development of Murine Depressive-Like Behavior. J. Immunol. 2009, 182, 3202–3212. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.W.; Norden, D.M.; Skendelas, J.P.; Huang, Y.; O’Connor, J.C.; Lawson, M.; Dantzer, R.; Kelley, K.W.; Godbout, J.P. Indoleamine 2,3-Dioxygenase Inhibition Attenuates Lipopolysaccharide Induced Persistent Microglial Activation and Depressive-Like Complications in Fractalkine Receptor (CX(3)CR1)-Deficient Mice. Brain Behav. Immun. 2013, 31, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Rharass, T.; Lantow, M.; Gbankoto, A.; Weiss, D.G.; Panáková, D.; Lucas, S. Ascorbic Acid Alters Cell Fate Commitment of Human Neural Progenitors in a WNT/β-Catenin/ROS Signaling Dependent Manner. J. Biomed. Sci. 2017, 16, 78. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, S.; Arcuri, C.; Hunot, S.; Mecca, C.; Codini, M.; Laurenti, M.E.; Ferri, I.; Loreti, E.; Garcia-Gil, M.; Traina, G.; et al. Effect of Vitamin D in HN9.10e Embryonic Hippocampal Cells and in Hippocampus from MPTP-Induced Parkinson’s Disease Mouse Model. Front. Cell. Neurosci. 2018, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Gooch, H.; Petty, A.; McGrath, J.J.; Eyles, D. Vitamin D and the Brain: Genomicand Non-Genomic Actions. Mol. Cell. Endocrinol. 2017, 453, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Haushalter, C.; Asselin, L.; Fraulob, V.; Dollé, P.; Rhinn, M. Retinoic Acid Controls Early Neurogenesis in the Developing Mouse Cerebral Cortex. Dev. Biol. 2017, 430, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lin, Y.; Lin, H.; Chen, Y.; Wang, H.; Shi, J. Application of Propyl Gallate Alleviates Pericarp Browning in Harvested Longan Fruit by Modulating Metabolisms of Respiration and Energy. Food Chem. 2018, 240, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.M.; Pickard, M.R.; Orme, R.P.; Hawkins, C.P.; Fricker, R.A. Nicotinamide Promotes Neuronal Differentiation of Mouse Embryonic Stem Cells in Vitro. Neuroreport 2013, 24, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.M.; Pickard, M.R.; Orme, R.P.; Hawkins, C.P.; Williams, A.C.; Fricker, R.A. Nicotinamide Alone Accelerates the Conversion of Mouse Embryonic Stem Cells into Mature Neuronal Populations. PLoS ONE 2017, 12, e0183358. [Google Scholar] [CrossRef] [PubMed]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The Metabolome Regulates the Epigenetic Landscape During Naive-to-Primed Human Embryonic Stem Cell Transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Lin, S.H.; Maiese, K. The NAD+ Precursor Nicotinamide Governs Neuronal Survival During Oxidative Stress Through Protein Kinase B Coupled to FOXO3a and Mitochondrial Membrane Potential. J. Cereb. Blood Flow Metab. 2004, 24, 728–743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xie, Y.; Wang, T.; Bi, J.; Li, H.; Zhang, L.Q.; Ye, S.Q.; Ding, S. Neuronal Protective Role of PBEF in a Mouse Model of Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1962–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, Q.; Bao, R.; Zhang, N.; Wang, Y.; Polo-Parada, L.; Tarim, A.; Alemifar, A.; Han, X.; Wilkins, H.M.; et al. Deletion of Nampt in Projection Neurons of Adult Mice Leads to Motor Dysfunction, Neurodegeneration, and Death. Cell Rep. 2017, 20, 2184–2200. [Google Scholar] [CrossRef] [PubMed]
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian Degeneration: An Emerging Axon Death Pathway Linking Injury and Disease. Nat. Rev. Neurosci. 2014, 15, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Loreto, A.; Di Stefano, M.; Gering, M.; Conforti, L. Wallerian Degeneration Is Executed by an NMN-SARM1-Dependent Late Ca2+ Influx butOnly Modestly Influenced by Mitochondria. Cell Rep. 2015, 13, 2539–2552. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.W.; Smith, C.B.; Schmidt, M.S.; Cambronne, X.A.; Cohen, M.S.; Migaud, M.E.; Brenner, C.; Goodman, R.H. Pharmacological Bypass of NAD(+) Salvage Pathway Protects Neurons from Chemotherapy-induced Degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 10654–10659. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased Nuclear NAD Biosynthesis and SIRT1 Activation Prevent Axonal Degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Gilley, J.; Coleman, M.P. Endogenous Nmnat2 Is an Essential Survivalfactor for Maintenance of Healthy Axons. PLoS Biol. 2010, 8, e1000300. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali, E. A Review on Alzheimer’s Disease Pathophysiology and Its Management: An Update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Scherr, P.A.; Tangney, C.C.; Hebert, L.E.; Bennett, D.A.; Wilson, R.S.; Aggarwal, N. Dietary Niacin and Risk of Incident Alzheimer’s Disease and of Cognitive Decline. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Schneider, J.A.; Tangney, C.C. Thoughts on B-vitamins and Dementia. J. Alzheimers Dis. 2006, 9, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.C.; Hill, L.J.; Ramsden, D.B. Nicotinamide, NAD(P)(H), and Methylgroup Homeostasis Evolved and Became a Determinant of Ageing Diseases: Hypotheses and Lessons from Pellagra. Curr. Gerontol. Geriatr. Res. 2012, 2012, 302875. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Pitta, M.; Jiang, H.; Le, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattson, M.P. Nicotinamide Forestalls Pathology and Cognitive Decline in Alzheimer Mice: Evidence for Improved Neuronal Bioenergetics and Autophagy Procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, X.; Yang, Y.; Takata, T.; Sakurai, T. Nicotinamide Mononucleotide Protects Against β-Amyloid Oligomer-Induced Cognitive Impairment and Neuronal Death. Brain Res. 2016, 1643, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Yang, S.J. Nicotinamide Reduces Amyloid Precursor Protein and Presenilin 1 in Brain Tissues of Amyloid Beta-Tail Vein Injected Mice. Clin. Nutr. Res. 2017, 6, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ Supplementation Normalizes Key Alzheimer’s Features and DNA Damage Responses in a New AD Mouse Model with Introduced DNA Repair Deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Wencel, P.L.; Lukiw, W.J.; Strosznajder, J.B.; Strosznajder, R.P. Inhibition of poly(ADP-ribose) Polymerase-1 Enhances Gene Expression of Selected Sirtuins and APP Cleaving Enzymes in Amyloid Beta Cytotoxicity. Mol. Neurobiol. 2018, 55, 4612–4623. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, C.; Trovato Salinaro, A.; Scuto, M.; Fronte, V.; Cambria, M.T.; Pennisi, M.; Bella, R.; Milone, P.; Graziano, A.; Crupi, R.; et al. Cellular Stress Response, Sirtuins and UCP Proteins in Alzheimer Disease: Role of Vitagenes. Immun. Ageing 2013, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, L.; Roriz-Cruz, M. Sirtuin 1 and Alzheimer’s Disease: An Up-To-Date Review. Neuropeptides 2018, 71, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, M.C.; Ali, Y.O.; Zhu, J.; Wu, C.S.; Oka, K.; Zhai, R.G.; Lu, H.C. CREB-Activity and NMNAT2 Transcription are Down-Regulated Prior to Neurodegeneration, while NMNAT2 Overexpression is Neuroprotective, in a Mouse Model of Human Tauopathy. Hum. Mol. Genet. 2012, 21, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.O.; Allen, H.M.; Yu, L.; Li-Kroeger, D.; Bakhshizadehmahmoudi, D.; Hatcher, A.; McCabe, C.; Xu, J.; Bjorklund, N.; Taglialatela, G.; et al. NMNAT2:HSP90 Complex Mediates Proteostasis in Proteinopathies. PLoS Biol. 2016, 14, e1002472. [Google Scholar] [CrossRef] [PubMed]
- Wakade, C.; Chong, R.; Bradley, E.; Thomas, B.; Morgan, J. Upregulation of GPR109A in Parkinson’s Disease. PLoS ONE 2014, 9, e109818. [Google Scholar] [CrossRef] [PubMed]
- Aaseth, J.; Dusek, P.; Roos, P.M. Prevention of Progression in Parkinson’s Disease. Biometals 2018, 31, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.W.; Bradbury, K.A.; Schneider, J.S. Broad Neuroprotective Profile of Nicotinamide in Different Mouse Models of MPTP-Induced Parkinsonism. Eur. J. Neurosci. 2008, 28, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Wakade, C.; Giri, B.; Malik, A.; Khodadadi, H.; Morgan, J.C.; Chong, R.K.; Baban, B. Niacin Modulates Macrophage Polarization in Parkinson’s Disease. J. Neuroimmunol. 2018, 320, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, J.; Sheng, R.; Li, M.; Wang, Y.; Han, R.; Han, F.; Chen, Z.; Qin, Z.H. Reduced Nicotinamide Adenine Dinucleotide Phosphate Inhibits MPTP-Induced Neuroinflammation and Neurotoxicity. Neuroscience 2018, 391, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Parsons, R.B.; Smith, M.L.; Williams, A.C.; Ramsden, D.B. Expression of Nicotinamide N-Methyltransferase (E.C. 2.1.1.1) in the Parkinsonian Brain. J. Neurol. Exp. Neurol. 2002, 61, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Parsons, R.B.; Smith, S.W.; Waring, R.H.; Williams, A.C.; Ramsden, D.B. High Expression of Nicotinamide N-Methyltransferase in Patients with Idiopathic Parkinson’s Disease. Neurosci. Lett. 2003, 342, 13–16. [Google Scholar] [CrossRef]
- Williams, A.C.; Cartwright, L.S.; Ramsden, D.B. Parkinson’s Disease: The First Common Neurological Disease due to Auto-Intoxication? QJM 2005, 98, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.G.; Saldanha, M.; Mistry, R.J.; Dexter, D.T.; Ramsden, D.B.; Parsons, R.B. Nicotinamide N-Methyltransferase Expression in SH-SY5Y Neuroblastoma and N27 Mesencephalic Neurones Induces Changes in Cell Morphology via Ephrin-B2 and Aktsignalling. Cell Death Dis. 2013, 4, 669. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Sharma, R.K.; Athilingam, T.; Sinha, P.; Sinha, N.; Thakur, A.K. NMR Spectroscopy-based Metabolomics of Drosophila Model of Huntington’s Disease Suggests Altered Cell Energetics. J. Proteome Res. 2017, 16, 3863–3872. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Feany, M.B. Comparison of Pathways Controlling Toxicity in the Eye and Brain in Drosophila Models of Human Neurodegenerative Diseases. Hum. Mol. Genet. 2004, 13, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Hathorn, T.; Snyder-Keller, A.; Messer, A. Nicotinamide Improves Motor Deficits and Upregulates PGC-1α and BDNF Gene Expression in a Mouse Model of Huntington’s Disease. Neurobiol. Dis. 2011, 41, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Diwan, V.; Kaur, H.; Bhateja, D.; Singh, C.K.; Sharma, S.; Padi, S.S.V. Nicotinamide Reverses Behavioral Impairments and Provides Neuroprotection in 3-Nitropropionic Acid Induced Animal Model of Huntington’s Disease: Implication of Oxidative Stress- Poly(ADP- Ribose) Polymerase Pathway. Metab. Brain Dis. 2018, 33, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Chidambaram, S.B.; Vijayan, R.; Sekar, S.; Mani, S.; Rajamani, B.; Ganapathy, R. Simultaneous Blockade of NMDA Receptors and PARP-1 Activity Synergistically Alleviate Immunoexcitotoxicity and Bioenergetics in 3-Nitropropionic Acid Intoxicated Mice: Evidences from Memantine and 3-Aminobenzamide Interventions. Eur. J. Pharmacol. 2017, 803, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Okuda, H.; Nishida, K.; Fujimoto, S.; Nagasawa, K. Protective Effect of Nicotinamide Against Poly(ADP-Ribose) Polymerase-1-Mediated Astrocyte Death Depends on Its Transporter-Mediated Uptake. Life Sci. 2010, 86, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Sadri-Vakili, G.; Cha, J.H. Histone Deacetylase Inhibitors: A Novel Therapeutic Approach to Huntington’s Disease (Complex Mechanism of Neuronal Death). Curr. Alzheimer Res. 2006, 3, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Pallos, J.; Bodai, L.; Lukacsovich, T.; Purcell, J.M.; Steffan, J.S.; Thompson, L.M.; Marsh, J.L. Inhibition of Specific HDACs and Sirtuins Suppresses Pathogenesis in a Drosophila Model of Huntington’s Disease. Hum. Mol. Genet. 2008, 17, 3767–3775. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD+ in Aging, Metabolism, and Neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, M.S.; Sinclair, D.A. Slowing Ageing by Design: The Rise of NAD. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Jęśko, H.; Wencel, P.; Strosznajder, R.P.; Strosznajder, J.B. Sirtuins and Their Roles in Brain Aging and Neurodegenerative Disorders. Neurochem. Res. 2017, 42, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.C.; Ramsden, D.B. Autotoxicity, Methylation and a Road to the Prevention of Parkinson’s Disease. J. Clin. Neurosci. 2005, 12, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Loh, S.H.; Martins, L.M. Enhancing NAD(+) Salvage Metabolism is Neuroprotective in a PINK1 Model of Parkinson’s Disease. Biol. Open 2017, 6, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Testa, C.M.; Jankovic, J. Huntington Disease: A Quarter Century of Progress Since the Gene Discovery. J. Neurol. Sci. 2019, 396, 52–68. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s Disease: A Clinical Review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Tulino, R.; Benjamin, A.C.; Jolinon, N.; Smith, D.L.; Chini, E.N.; Carnemolla, A.; Bates, G.P. SIRT1 Activity Is Linked to Its Brain Region-Specific Phosphorylation and Is Impaired in Huntington’s Disease Mice. PLoS ONE 2016, 11, e0145425. [Google Scholar] [CrossRef] [Green Version]
- Naia, L.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira-Sousa, S.I.; Caldeira, G.L.; Carmo, C.; Laço, M.N.; Hayden, M.R.; Oliveira, C.R.; Rego, A.C. Comparative Mitochondrial-Based Protective Effects of Resveratrol and Nicotinamide in Huntington’s Disease Models. Mol. Neurobiol. 2017, 54, 5385–5399. [Google Scholar] [CrossRef] [PubMed]
- Harrison, I.F.; Powell, N.M.; Dexter, D.T. The Histone Deacetylase Inhibitor Nicotinamide Exacerbates Neurodegeneration in the Lactacystin Rat Model of Parkinson’s Disease. J. Neurochem. 2019, 148, 136–156. [Google Scholar] [CrossRef] [PubMed]
- Hoane, M.R.; Akstulewicz, S.L.; Toppen, J. Treatment with Vitamin B3 Improves Functional Recovery and Reduces GFAP Expression Following Traumatic Brain Injury in Rats. J. Neurotrauma 2003, 20, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Goffus, A.M.; Anderson, G.D.; Hoane, M. Sustained Delivery of Nicotinamide Limits Cortical Injury and Improves Functional Recovery Following Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2010, 3, 145–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonder Haar, C.; Maas, W.R.; Jacobs, E.A.; Hoane, M.R. Deficits in Discrimination after Experimental Frontal Brain Injury Are Mediated by Motivation and Can Be Improved by Nicotinamide Administration. J. Neurotrauma 2014, 31, 1711–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonder Haar, C.; Anderson, G.D.; Hoane, M.R. Continuous Nicotinamide Administration Improves Behavioral Recovery and Reduces Lesion Size Following Bilateral Frontal Controlled Cortical Impact Injury. Behav. Brain Res. 2011, 224, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Choi, B.Y.; Yoo, B.H.; Sohn, M.; Ying, W.; Swanson, R.A.; Suh, S.W. Prevention of Traumatic Brain Injury-Induced Neuron Death by Intranasal Delivery of Nicotinamide Adenine Dinucleotide. J. Neurotrauma 2012, 29, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Swan, A.A.; Chandrashekar, R.; Beare, J.; Hoane, M.R. Preclinical Efficacy Testing in Middle-Aged Rats: Nicotinamide, a Novel Neuroprotectant, Demonstrates Diminished Preclinical Efficacy after Controlled Cortical Impact. J. Neurotrauma 2011, 28, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, W.Y.; Suh, G.J.; Kim, K.S.; Lee, H.J.; Jeong, K.Y.; Kwak, Y.H.; Kim, K. Niacin Suppresses the Mitogen-Activated Protein Kinase Pathway and Attenuates Brain Injury after Cardiac Arrest in Rats. Crit. Care Med. 2013, 41, e223–e232. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide Mononucleotide Inhibits Post-Ischemic NAD(+) Degradation and Dramatically Ameliorates Brain Damage Following Global Cerebral Ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Shetty, P.K.; Galeffi, F.; Turner, D.A. Nicotinamide Pre-treatment Ameliorates NAD(H) Hyperoxidation and Improves Neuronal Function after Severe Hypoxia. Neurobiol. Dis. 2014, 62, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cui, X.; Zacharek, A.; Jiang, H.; Roberts, C.; Zhang, C.; Lu, M.; Kapke, A.; Feldkamp, C.S.; Chopp, M. Niaspan Increases Angiogenesis and Improves Functional Recovery after Stroke. Ann. Neurol. 2007, 62, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cui, X.; Zacharek, A.; Ding, G.L.; Shehadah, A.; Jiang, Q.; Lu, M.; Chopp, M. Niaspan Treatment Increases Tumor Necrosis Factor-Alpha-Converting Enzyme and Promotes Arteriogenesis after Stroke. J. Cereb. Blood Flow Metab. 2009, 29, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Kwon, W.Y.; Suh, G.J.; Kim, K.S.; Jung, Y.S.; Kim, S.H.; Lee, A.R.; You, K.M.; Park, M.J. Niacin and Selenium Attenuate Brain Injury After Cardiac Arrest in Rats by Up-Regulating DJ-1-Akt Signaling. Crit. Care Med. 2018, 46, e788–e796. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.C.; Anderson, G.D.; Kantor, E.D.; Hoane, M.R. A Comparison of the Effects of Nicotinamide and Progesterone on Functional Recovery of Cognitive Behavior Following Cortical Contusion Injury in the Rat. J. Neurotrauma 2012, 29, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.C.; Hoane, M.R.; McConomy, K.S.; Farin, F.M.; Bammler, T.K.; MacDonald, J.W.; Kantor, E.D.; Anderson, G.D. A Combination Therapy of Nicotinamide and Progesterone Improves Functional Recovery following Traumatic Brain Injury. J. Neurotrauma 2015, 32, 765–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Xu, T.Y.; Guan, Y.F.; Tian, W.W.; Viollet, B.; Rui, Y.C.; Zhai, Q.W.; Su, D.F.; Miao, C.Y. Nicotinamide Phosphoribosyltransferase Protects against Ischemic Stroke through SIRT1-Dependent Adenosine Monophosphate-Activated Kinase Pathway. Ann. Neurol. 2011, 69, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kraus, W.L. SIRT1-Dependent Regulation of Chromatin and Transcription: Linking NAD(+) Metabolism and Signaling to the Control of Cellular Functions. Biochim. Biophys. Acta 2010, 1804, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.; Xing, J.; Chen, X.; Stetler, R.A.; Wen, Z.; Gan, Y.; Zhang, F.; Gao, Y.; Chen, J.; Leak, R.K.; et al. Neuronal NAMPT is Released after Cerebral Ischemia and Protects against White Matter Injury. J. Cereb. Blood Flow Metab. 2014, 34, 1613–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardison, W.R.; Yorns, H.H., Jr. Mitochondrial Dysfunction in Migraine. Semin. Pediatr. Neurol. 2013, 20, 188–193. [Google Scholar]
- Neri, M.; Frustaci, A.; Milic, M.; Valdiglesias, V.; Fini, M.; Bonassi, S.; Barbanti, P. A Meta-Analysis of Biomarkers Related to Oxidative Stress and Nitric Oxide Pathway in Migraine. Cephalalgia 2015, 35, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Bohár, Z.; Párdutz, Á.; Vécsei, L. Tryptophan Catabolites and Migraine. Curr. Pharm. Des. 2016, 22, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Awad, J.A.; Oates, J.A.; Roberts, L.J. Identification of Skin as a Major Site on Prostaglandin D2 Release Following Oral Administration of Niacin in Humans. J. Investig. Dermatol. 1992, 98, 812–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, J.D.; Parsons, W.G.; Roberts, L., II. Release of Markedly Increased Quantities of Prostaglandin D2 in Vivo in Humans Following the Administration of Nicotinic Acid. Prostaglandins 1989, 38, 263–274. [Google Scholar] [CrossRef]
- Kim, E.J.; Lim, S.Y.; Lee, H.J.; Lee, J.Y.; Choi, S.; Kim, S.Y.; Kim, J.M.; Shin, I.S.; Yoon, J.S.; Yang, S.J.; et al. Low Dietary Intake of n-3 Fatty Acids, Niacin, Folate, and Vitamin C in Korean Patients with Schizophrenia and the Development of Dietary Guidelines for Schizophrenia. Nutr. Res. 2017, 45, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Sun, X.Y.; Zhang, C.B.; Yan, J.J.; Zhao, Q.Q.; Yang, S.Y.; Yan, L.L.; Huang, N.H.; Zeng, J.; Liao, J.Y.; et al. Association Between B Vitamins and Schizophrenia: A Population-Based Case-Control Study. Psychiatry Res. 2018, 259, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, B.H. Nicotinic Acid Long-Term Effectiveness in a Patient with Bipolar Type II Disorder: A Case of Vitamin Dependency. Nutrients 2018, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Ryou, M.G.; Mallet, R.T. An In Vitro Oxygen-Glucose Deprivation Model for Studying Ischemia-Reperfusion Injury of Neuronal Cells. Methods Mol. Biol. 2018, 1717, 229–235. [Google Scholar] [CrossRef]
- Martin, E.; Rosenthal, R.E.; Fiskum, G. Pyruvate Dehydrogenase Complex: Metabolic Link to Ischemic Brain Injury and Target of Oxidative Stress. J. Neurosci. Res. 2005, 79, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Mokudai, T.; Ayoub, I.A.; Sakakibara, Y.; Lee, E.J.; Ogilvy, C.S.; Maynard, K.I. Delayed Treatment with Nicotinamide (Vitamin B3) Improves Neurological Outcome and Reduces Infarct Volume after Transient Focal Cerebral Ischemia in Wistar Rats. Stroke 2000, 31, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, I.A.; Lee, E.J.; Ogilvy, C.S.; Beal, M.F.; Maynard, K.I. Nicotinamide Reduces Infarction up to Two Hours after the Onset of Permanent Focal Ischemia in Wistar Rats. Neurosci. Lett. 1999, 259, 21–24. [Google Scholar] [CrossRef]
- Sakakibara, Y.; Mitha, A.P.; Ogilvy, C.S.; Maynard, K.I. Post-Treatment with Nicotinamide (Vitamin B3) Reduces the Infarct Volume Following Permanent Focal Ischemia in Female Sprague–Dawley and Wistar Rats. Neurosci. Lett. 2000, 281, 111–114. [Google Scholar] [CrossRef]
- Sakakibara, Y.; Mitha, A.P.; Ayoub, I.A.; Ogilvy, C.S.; Maynard, K.I. Delayed Treatment with Nicotinamide (Vitamin B3) Reduces the Infarct Volume Following Focal Cerebral Ischemia in Spontaneously Hypertensive Rats, Diabetic and Non-Diabetic Fischer 344 Rats. Brain Res. 2002, 931, 68–73. [Google Scholar] [CrossRef]
- Feng, Y.; Paul, I.A.; LeBlanc, M.H. Nicotinamide Reduces Hypoxic Ischemic Brain Injury in the Newborn Rat. Brain Res. Bull. 2006, 69, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, P.; Mullally, W.J. Headache. Am. J. Med. 2018, 131, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Close Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders: 3rd Edition. Cephalalgia 2013, 33, 629–808. [Google Scholar] [CrossRef] [PubMed]
- Nattagh-Eshtivani, E.; Sani, M.A.; Dahri, M.; Ghalichi, F.; Ghavami, A.; Arjang, P.; Tarighat-Esfanjani, A. The Role of Nutrients in the Pathogenesis and Treatment of Migraine Headaches: Review. Biomed. Pharmacother. 2018, 102, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Goldzieher, J.W.; Popkin, G.L. Treatment of Headache with Intravenous Sodium Nicotinate. J. Am. Med. Assoc. 1946, 131, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Grenfell, R.F. Treatment of Migraine with Nicotinic Acid. Am. Pract Dig. Treat. 1949, 3, 542–544. [Google Scholar] [PubMed]
- Grenfell, R.F. Treatment of Tension Headache. Am. Pract. Dig. Treat. 1951, 2, 933–936. [Google Scholar] [PubMed]
- Morgan, Z.R. Nicotinic Acid Therapy in Vasoconstriction Type of Headache. Md. State Med. J. 1953, 2, 377–382. [Google Scholar] [PubMed]
- Morgan, Z.R. A Newer Method of Nicotinic Acid Therapy in Headache of the Vasoconstrictive Type. J. Am. Geriatr. Soc. 1955, 3, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Prousky, J.; Seely, D. The Treatment of Migraines and Tension-Type Headaches with Intravenous and Oral Niacin (Nicotinic Acid): Systematic Review of the Literature. Nutr. J. 2005, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.F. Tryptophan Kynurenine Metabolism as a Common Mediator of Genetic and Environmental Impacts in Major Depressive Disorder: The Serotonin Hypothesis Revisited 40 Years Later. Isr. J. Psychiatry Relat. Sci. 2010, 47, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M.; Schwarz, M.J.; Müller, N. The Role of the Kynurenine Metabolism in Major Depression. J. Neural Transm. 2012, 119, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Wonodi, I.; Schwarcz, R. Cortical Kynurenine Pathway Metabolism: A Novel Target for Cognitive Enhancement in Schizophrenia. Schizophr. Bull. 2010, 36, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.L.; Llenos, I.C.; Cwik, M.; Walkup, J.; Weis, S. Alterations in Kynurenine Precursor and Product Levels in Schizophrenia and Bipolar Disorder. Neurochem. Int. 2008, 52, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.; Drevets, W.C.; Smith, C.M.; Victor, T.A.; Wurfel, B.E.; Bellgowan, P.S.; Bodurka, J.; Teague, T.K.; Dantzer, R. Putative Neuroprotective and Neurotoxic Kynurenine Pathway Metabolites are Associated with Hippocampal and Amygdalar Volumes in Subjects with Major Depressive Disorder. Neuropsychopharmacology 2015, 40, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Funakoshi, H.; Takahashi, H.; Hayakawa, T.; Mizuno, S.; Matsumoto, K.; Nakamura, T. Tryptophan 2,3-Dioxygenase is a Key Modulator of Physiological Neurogenesis and Anxiety-Related Behavior in Mice. Mol. Brain 2009, 2, 8. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effector | Main Findings | Ref. | |
|---|---|---|---|
| Alzheimer’s disease | Niacin | Inverse association between AD and dietary niacin intakes | [135] |
| NAD+ | High brain levels restore mitochondrial function and antagonize cognitive decline | [138,139] | |
| Nam/Nam mononucleotide | Protect against Aβ-induced neurotoxicity via reduction of APP and PSEN-1 expression and ROS levels | [140,141] | |
| Nam riboside | Reduces DNA damage, neuroinflammation and cell death of hippocampal neurons | [142] | |
| SIRT1 | Supports the non-amyloidogenic pathway of AD Lessens AD neuroinflammation, oxidative stress and mitochondrial dysfunction | [143] [144,145] | |
| NMNAT1-3 | Protects against axon degeneration via reduction of nicotinamide mononucleotide levels and SIRT1 activation | [132,133] | |
| NMNAT2 | Activity downregulated prior to neurodegeneration; restoration of activity is neuroprotective against tauopathy Low gene expression in AD patients | [146] [147] | |
| Parkinson’s disease | Niacin | Increased intake enhances striatal dopamine synthesis and restores optimal NAD+/NADH ratio High levels sequester transition metal ions Low doses impact macrophage polarization from M1 (pro-inflammatory) to M2 (anti-inflammatory) profile | [148] [149,150] [151] |
| NAD+ | Decreased levels in PD patients | [148] | |
| NADPH | Inhibits MPTP+-induced oxidative stress and glia-mediated neuroinflammation | [152] | |
| NNMT | High levels in the cerebrospinal fluid and midbrain dopamine neurons of PD patients High activity associated with low activity of mitochondrial complex 1; it counteracts the MPP+-dependent toxicity on mitochondrial complex 1 and activates neuronal autophagy Induces neurite branching, synaptophysin expression and dopamine release | [153,154] [154,155] [156] | |
| Huntington’s disease | NAD | Low levels correlate with disease progression in Drosophila HD model | [157] |
| Nam | Protects against the toxicity of polyQ proteins in Drosophila HD models Restores BDNF protein levels, increases acetylated PGC-1α, improves motor deficits Prevents motor abnormality via PARP-1-dependent inhibition of neuronal death and oxidative stress | [158] [159] [160,161,162] | |
| SIRT1 | Rescues neurons from mutant huntingtin toxicity Ameliorates pathological mechanisms underlying disease onset | [163,164] |
| Effector | Main Findings | Ref. | |
|---|---|---|---|
| Ischemic and traumatic injuries | Niacin | Diminishes TBI-dependent behavioral deficits and improves functional recovery | [175,176,177,178,179,180] |
| Nam | Reduces neurologic deficits, hippocampal apoptosis, axonal injury and microglial activation in corpus callosum and oxidative stress; restores NAD(P) content; represses MAPK signaling and caspase 3 cleavage | [181] | |
| Nam mononucleotide | Ameliorates hippocampal injury and improves neurological outcome, by decreasing poly-ADP-ribosylated proteins and NAD+ catabolism | [182] | |
| Nam/PARP-1 antagonists | Pre-treatment improves ATP content and neuronal recovery during re-oxygenation | [183] | |
| Niaspan (niacin) | Increases local cerebral blood flow; promotes angiogenesis via angpt/Tie2, Akt and eNOS pathways; promotes arteriogenesis via TACE and Notch signaling; ameliorates functional deficits | [184,185] | |
| Niacin plus selenium | Attenuate cortical cell injury, via an increase in Akt phosphorylation and expression of Nrf2; reduce oxidative stress. | [186] | |
| Nam plus progesterone | Increase function recovery; reduce lesion cavitation and tissue loss; modulate expression of inflammatory and immune genes | [187,188] | |
| NAMPT | Decreased activity exacerbates post-ischemic brain damage Heterozygous gene deletion aggravates brain damage following photothrombosis-induced focal ischemia Gene over-expression reduces infarct size | [189,190] [190] [191] | |
| Headaches | Niacin | Restores mitochondrial energy metabolism Ameliorates blood flow and oxygenation in contracted skeletal muscle | [192,193] |
| Nicotinic acid | Dilates intracranial vessels and contracts extracranial vessels; increases skin biosynthesis of prostaglandin D2; rises plasma content of 9a,11b-prostaglandin F2 | [194,195,196] | |
| Psychiatric disorders | Niacin | Low dietary intakes in neuropsychiatric patients | [197] |
| Nam | Positive correlation between vitamin levels and schizophrenia Chronic supplementation effective in maintaining a bipolar type II patient stable and calm | [198] [199] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gasperi, V.; Sibilano, M.; Savini, I.; Catani, M.V. Niacin in the Central Nervous System: An Update of Biological Aspects and Clinical Applications. Int. J. Mol. Sci. 2019, 20, 974. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040974
Gasperi V, Sibilano M, Savini I, Catani MV. Niacin in the Central Nervous System: An Update of Biological Aspects and Clinical Applications. International Journal of Molecular Sciences. 2019; 20(4):974. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040974
Chicago/Turabian StyleGasperi, Valeria, Matteo Sibilano, Isabella Savini, and Maria Valeria Catani. 2019. "Niacin in the Central Nervous System: An Update of Biological Aspects and Clinical Applications" International Journal of Molecular Sciences 20, no. 4: 974. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040974