Role of Platelet Glycoprotein VI and Tyrosine Kinase Syk in Thrombus Formation on Collagen-Like Surfaces

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
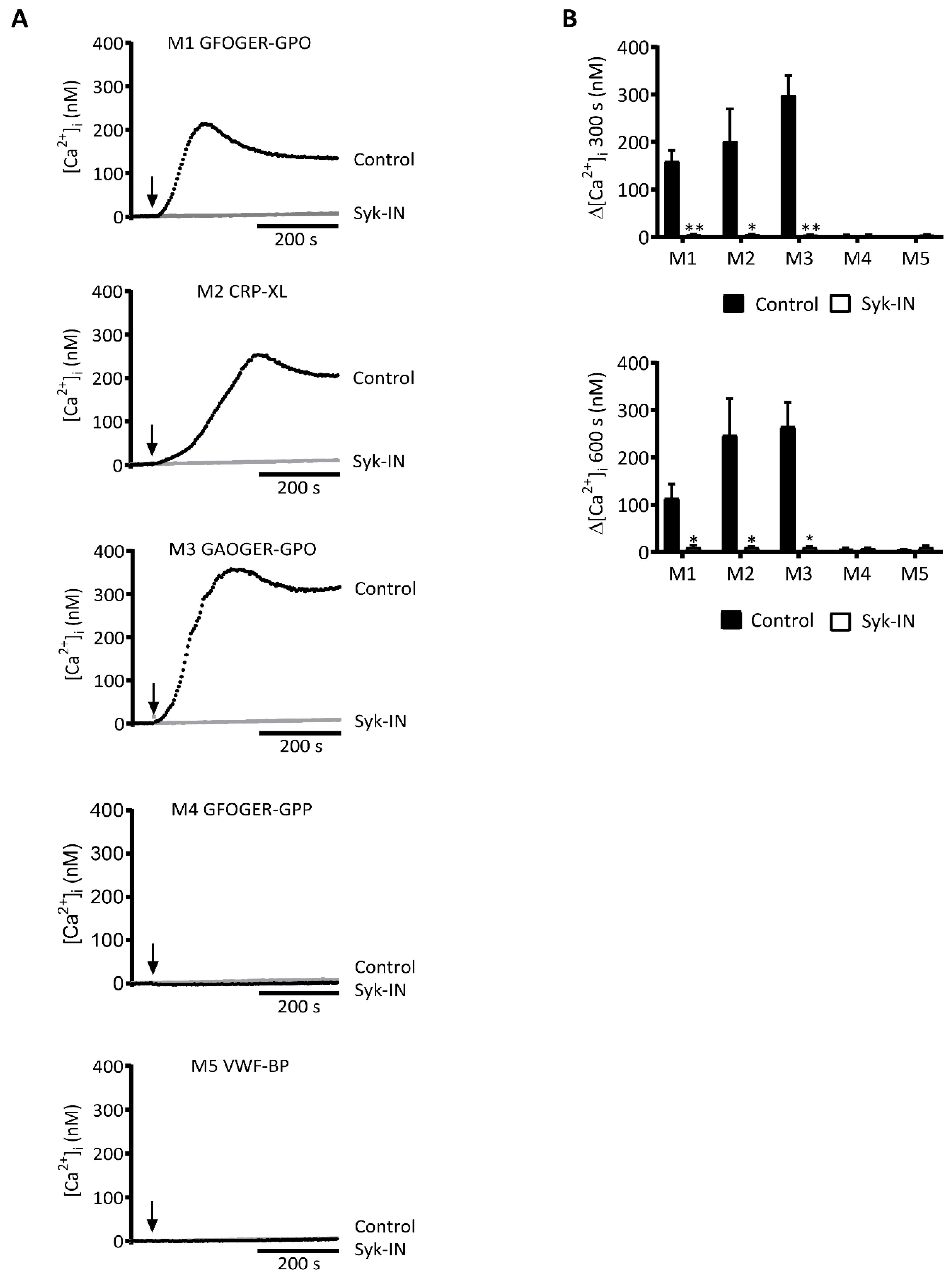
2.1. GPVI-Dependent and Syk-Dependent Platelet Activation by Collagen Peptides
2.2. GPVI- and Syk-Dependent Parameters of Thrombus Formation on Collagen Peptides
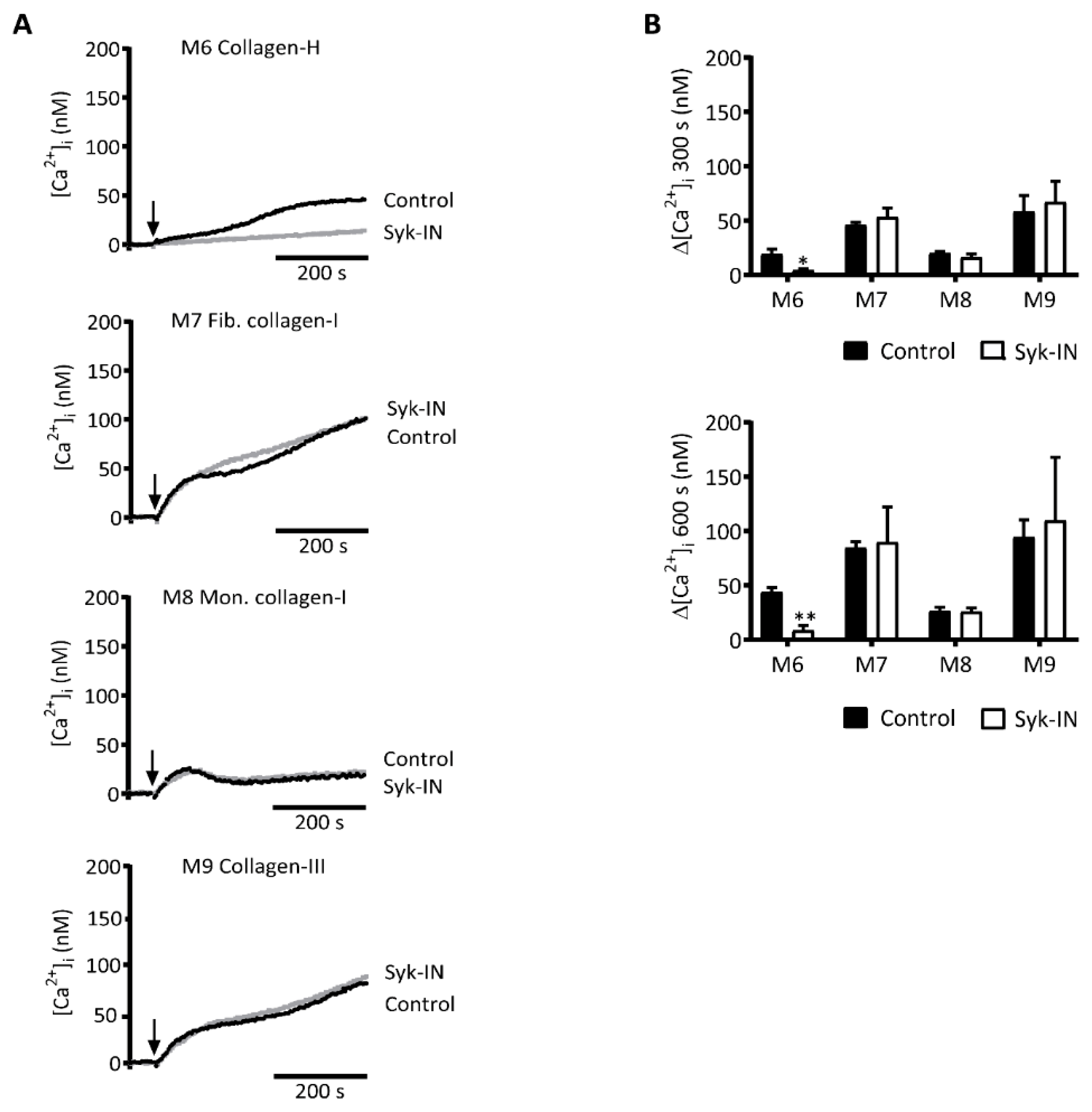
2.3. GPVI-Induced and Syk-Dependent Platelet Activation by Different Collagens
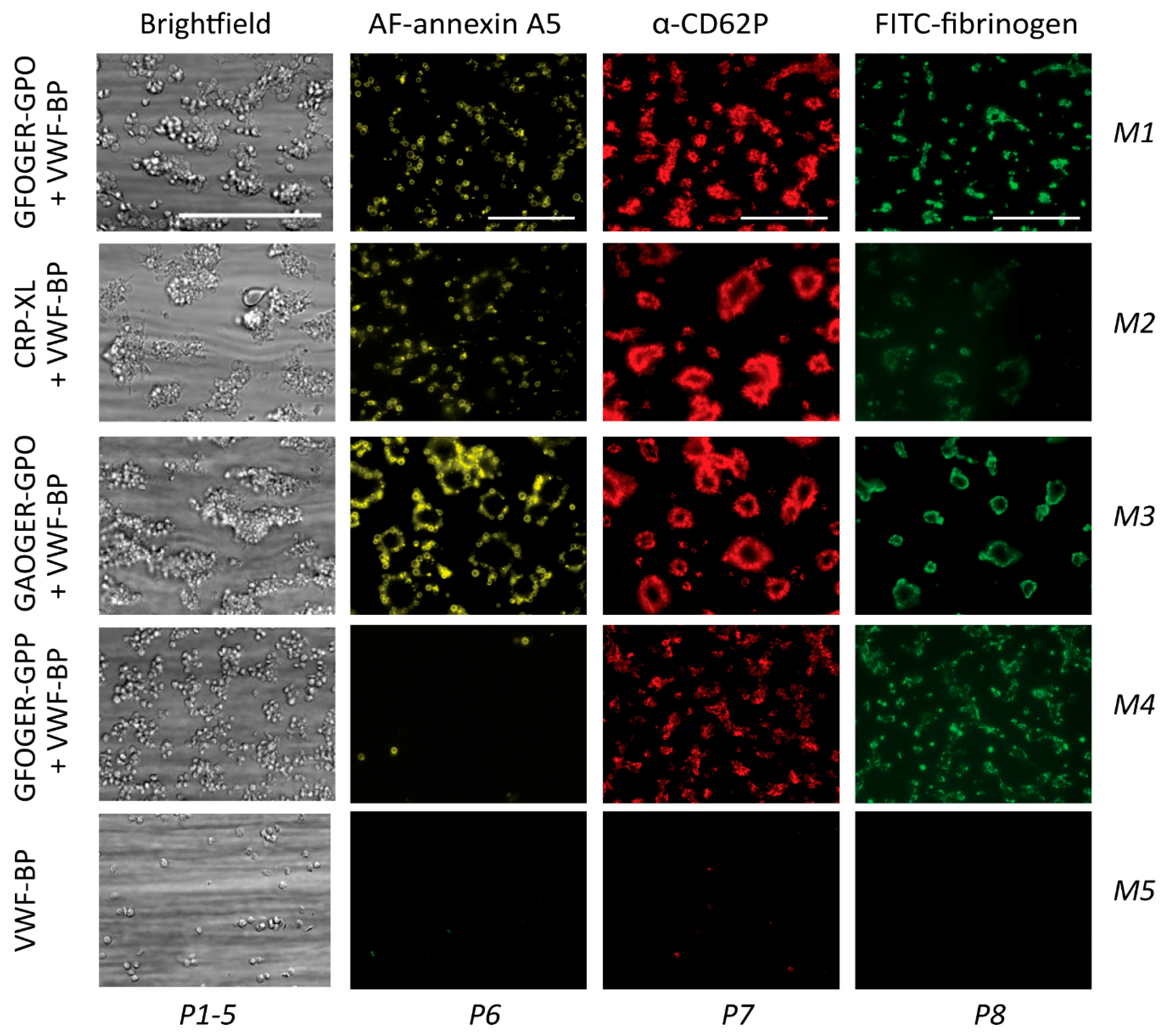
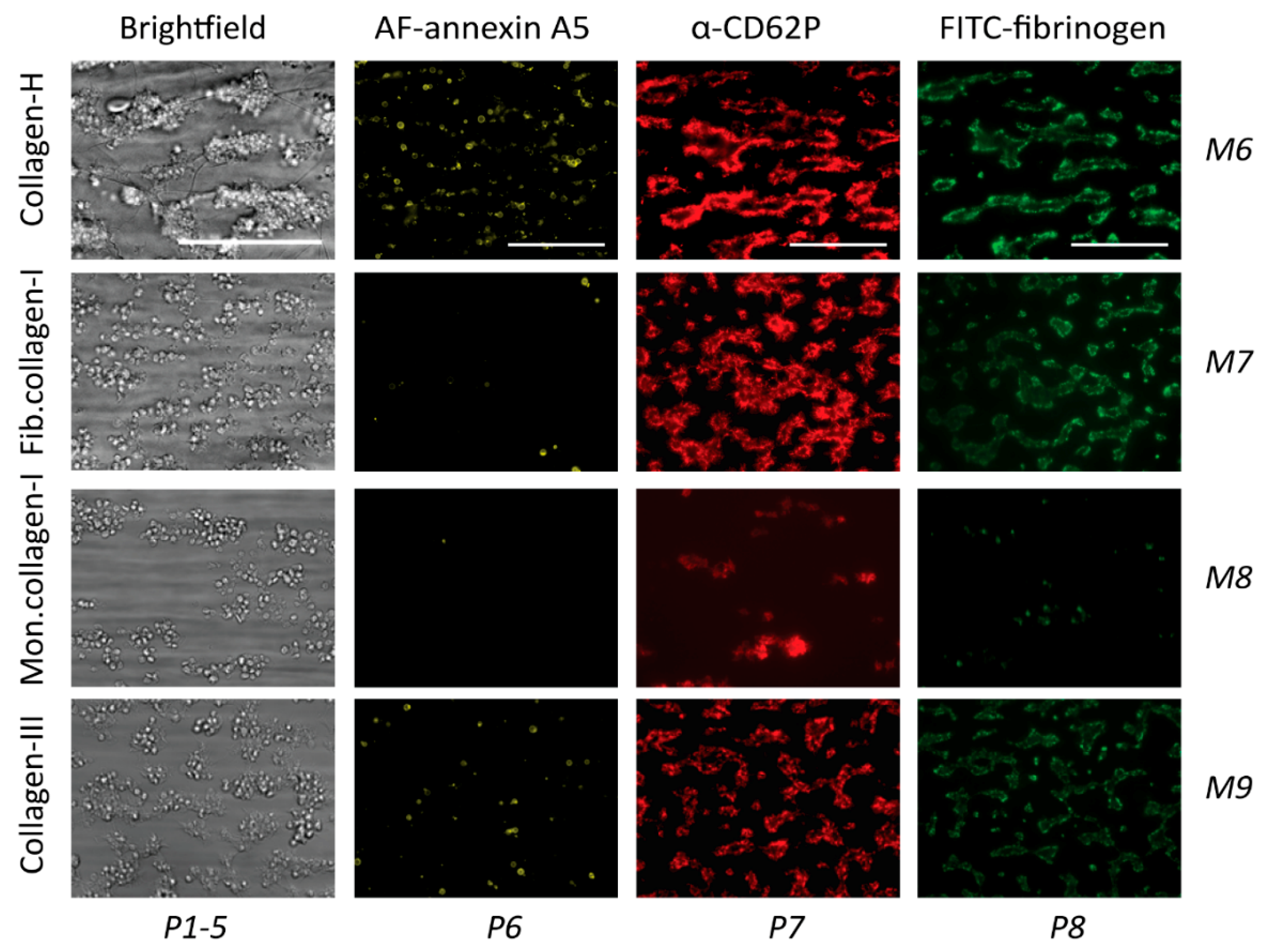
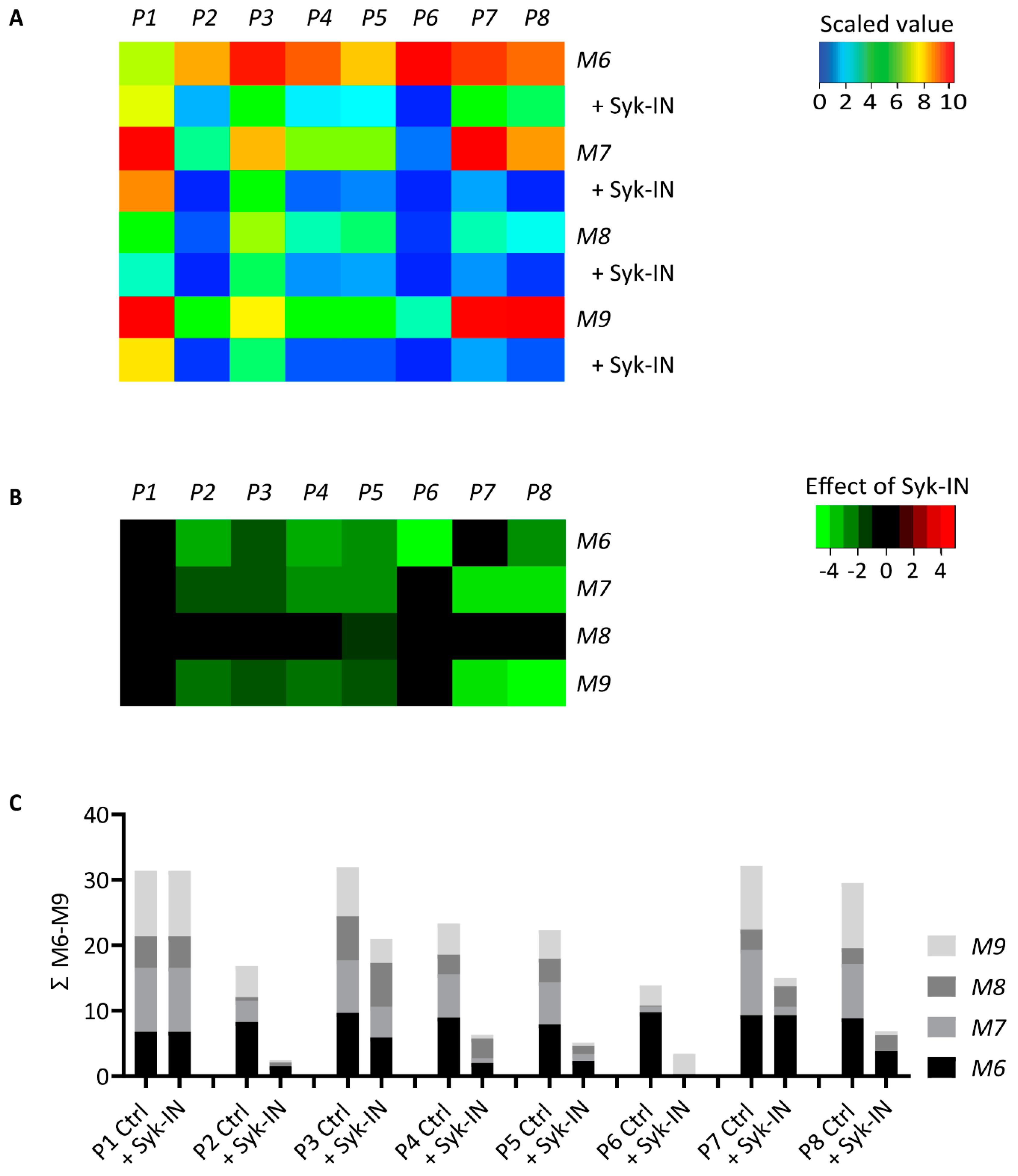
2.4. GPVI- and Syk-Dependent Platelet Responses in Thrombus Formation on Collagens
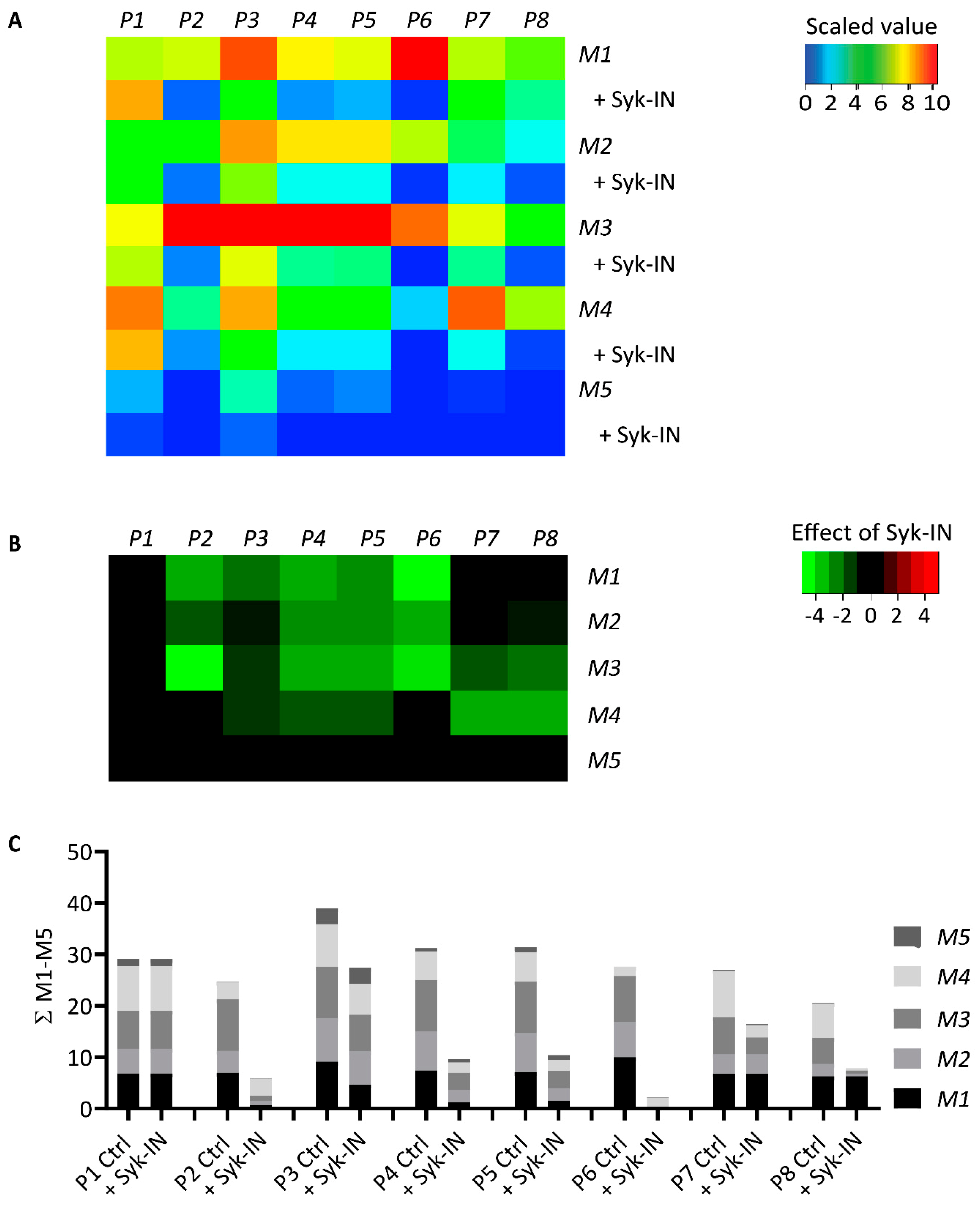
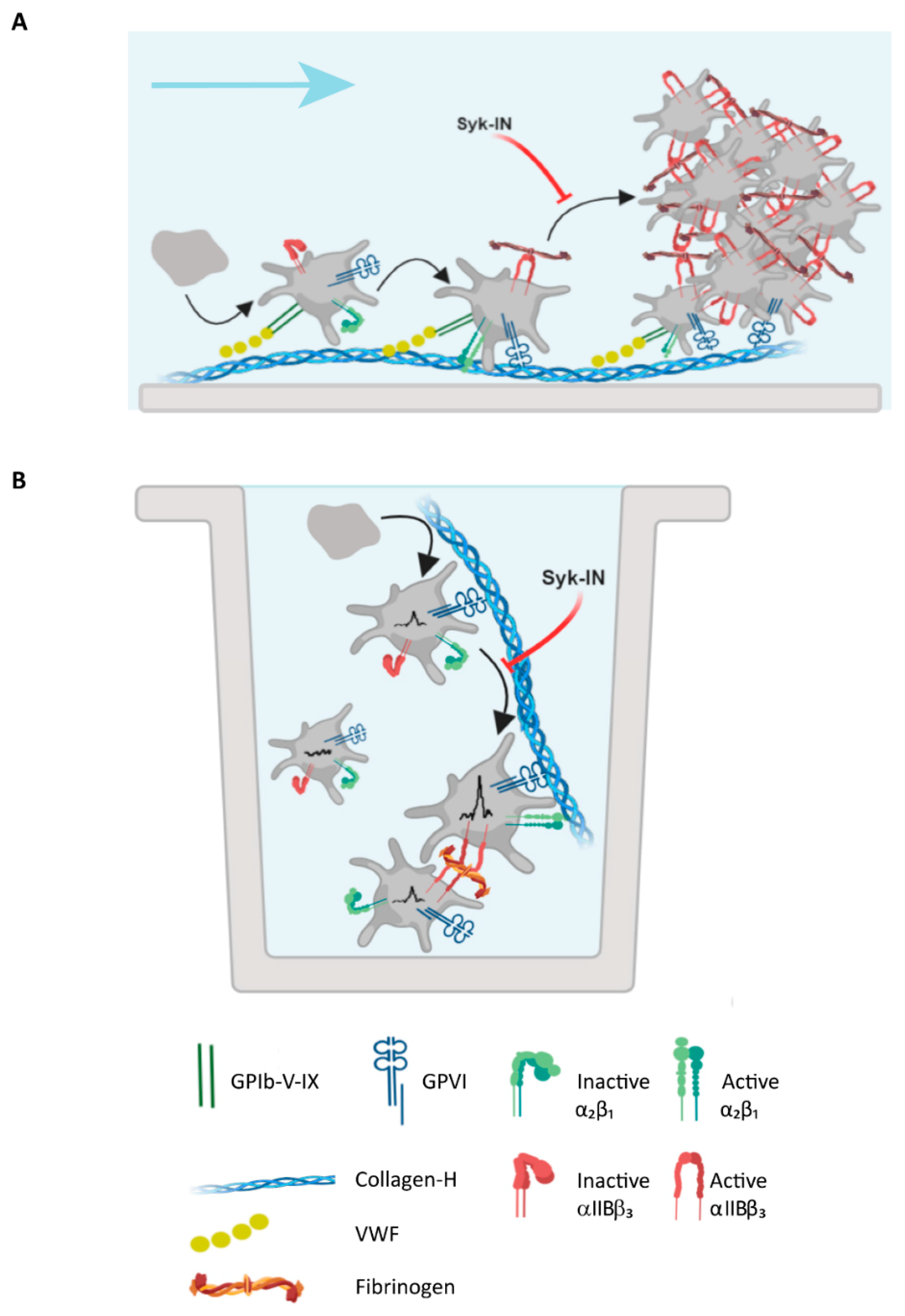
2.5. Modeling of the Role of GPVI in Thrombus Formation on Various Collagens
3. Discussion
3.1. Collagen Peptides and GPVI-Dependent Platelet Activation
3.2. Collagens and GPVI-Dependent Platelet Activation
3.3. Comparative Roles of GPVI and Syk in Platelet Activation
3.4. Conclusion
4. Materials and Methods
4.1. Materials
4.2. Blood Isolation
4.3. Platelet Isolation and Loading with Fura-2
4.4. Light Transmission Aggregometry
4.5. Whole-Blood Microfluidic Perfusion Over Microspots
4.6. Bright-Field and Fluorescence Microscopy
4.7. Cytosolic Ca2+ Measurements
4.8. Data Handling and Statistics
4.9. Modeling to Predict GPVI Activity
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.S. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef]
- Van der Meijden, P.E.; Heemskerk, J.W. Platelet biology and functions: New concepts and future clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Savage, B.; Almus-Jacobs, F.; Ruggeri, Z.M. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell 1998, 94, 657–666. [Google Scholar] [CrossRef]
- Atkinson, B.T.; Jarvis, G.E.; Watson, S.P. Activation of GPVI by collagen is regulated by a2b1 and secondary mediators. J. Thromb. Haemost. 2003, 1, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Auger, J.M.; Kuijpers, M.J.; Senis, Y.A.; Watson, S.P.; Heemskerk, J.W. Adhesion of human and mouse platelets to collagen under shear: A unifying model. FASEB J. 2005, 19, 825–827. [Google Scholar] [CrossRef] [PubMed]
- Kehrel, B.; Wierwille, S.; Clemetson, K.J.; Anders, O.; Steiner, M.; Knight, C.G.; Farndale, R.W.; Okuma, M.; Barnes, M.J. Glycoprotein VI is a major collagen receptor for platelet activation: It recognizes the platelet-activating quaternary structure of collagen, whereas CD36, glycoprotein IIb/IIIa, and von Willebrand factor do not. Blood 1998, 91, 491–499. [Google Scholar] [PubMed]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Baaten, C.C.; Meacham, S.; de Witt, S.M.; Feijge, M.A.; Adams, D.J.; Akkerman, J.W.; Cosemans, J.M.; Grassi, L.; Jupe, S.; Kostadima, M.; et al. A synthesis approach of mouse studies to identify genes and proteins in arterial thrombosis and bleeding. Blood 2018, 132, e35–e46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Witt, S.M.; Swieringa, F.; Cavill, R.; Lamers, M.M.; van Kruchten, R.; Mastenbroek, T.; Baaten, C.; Coort, S.; Pugh, N.; Schulz, A.; et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat. Commun. 2014, 5, 4257. [Google Scholar] [CrossRef] [Green Version]
- Pugh, N.; Simpson, A.M.; Smethurst, P.A.; de Groot, P.G.; Raynal, N.; Farndale, R.W. Synergism between platelet collagen receptors defined using receptor-specific collagen-mimetic peptide substrata in flowing blood. Blood 2010, 115, 5069–5079. [Google Scholar] [CrossRef]
- Siljander, P.R.; Munnix, I.C.; Smethurst, P.A.; Deckmyn, H.; Lindhout, T.; Ouwehand, W.H.; Farndale, R.W.; Heemskerk, J.W. Platelet receptor interplay regulates collagen-induced thrombus formation in flowing human blood. Blood 2004, 103, 1333–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munnix, I.C.; Kuijpers, M.J.; Auger, J.M.; Thomassen, C.M.; Panizzi, P.; van Zandvoort, M.A.; Rosing, J.; Bock, P.E.; Watson, S.P.; Heemskerk, J.W. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation. Regulation by transient integrin activation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2484–2490. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.; de Witt, S.; Solecka, B.A.; Kröning, M.; Obser, T.; Cosemans, J.M.E.M.; Schneppenheim, R.; Heemskerk, J.W.M.; Kannicht, C. Distinct role of von Willebrand factor triplet bands in glycoprotein Ib-dependent platelet adhesion and thrombus formation under flow. Semin. Thromb. Hemost. 2013, 39, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.G.; Morton, L.F.; Onley, D.J.; Peachey, A.R.; Ichinohe, T.; Okuma, M.; Farndale, R.W.; Barnes, M.J. Collagen-platelet interaction: Gly-Pro-Hyp is uniquely specific for platelet GPVI and mediates platelet activation by collagen. Cardiovasc. Res. 1999, 41, 450–457. [Google Scholar] [CrossRef]
- Smethurst, P.A.; Onley, D.J.; Jarvis, G.E.; O’Connor, M.N.; Knight, C.G.; Herr, A.B.; Ouwehand, W.H.; Farndale, R.W. Structural basis for the platelet-collagen interaction. The smallest motif within collagen that recognizes and activates platelet glycoprotein VI contains to glycine-proline-hydroxyproline triplets. J. Biol. Chem. 2007, 282, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Munnix, I.C.; Gilio, K.; Siljander, P.R.; Raynal, N.; Feijge, M.A.; Hackeng, T.M.; Deckmyn, H.; Smethurst, P.A.; Farndale, R.W.; Heemskerk, J.W. Collagen-mimetic peptides mediate flow-dependent thrombus formation by high- or low-affinity binding of integrin a2b1 and glycoprotein VI. J. Thromb. Haemost. 2008, 6, 2132–2142. [Google Scholar] [CrossRef]
- Siljander, P.R.; Hamaia, S.; Peachey, A.R.; Slatter, D.A.; Smethurst, P.A.; Ouwehand, W.H.; Knight, C.G.; Farndale, R.W. Integrin activation state determines selectivity for novel recognition sites in fibrillar collagens. J. Biol. Chem. 2004, 279, 47763–47772. [Google Scholar] [CrossRef]
- Ichinohe, T.; Takayama, H.; Ezumi, Y.; Arai, M.; Yamamoto, N.; Takahashi, H.; Okuma, M. Collagen-stimulated activation of Syk but not c-Src is severely compromised in human platelets lacking membrane glycoprotein VI. J. Biol. Chem. 1997, 272, 63–68. [Google Scholar] [CrossRef]
- Melford, S.K.; Turner, M.; Briddon, S.J.; Tybulewicz, V.L.; Watson, P.S. Syk and Fyn are required by mouse megakaryocytes for the rise in intracellular calcium induced by a collagen-related peptide. J. Biol. Chem. 1997, 272, 27539–27542. [Google Scholar] [CrossRef]
- Watson, S.P.; Herbert, J.M.J.; Pollitt, A.Y. GPVI and CLEC-2 in hemostasis and vascular integrity. J. Thromb. Haemost. 2010, 8, 1457–1467. [Google Scholar] [CrossRef]
- Badolia, R.; Kostyak, J.C.; Dangelmaier, C.; Kunapuli, S.P. Syk activity is dispensable for platelet GPIb-IX-V signaling. Int. J. Mol. Sci. 2017, 18, 1238. [Google Scholar] [CrossRef] [PubMed]
- Yanaga, F.; Poole, A.; Asselin, J.; Blake, R.; Schieven, G.L.; Clark, E.A.; Law, C.L.; Watson, S.P. Syk interacts with tyrosine-phosphorylated proteins in human platelets activated by collagen and cross-linking of the FcgIIA receptor. Biochem. J. 1995, 311, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Munnix, I.C.; Strehl, A.; Kuijpers, M.J.; Auger, J.M.; van der Meijden, P.E.; van Zandvoort, M.A.; oude Egbrink, M.; Nieswandt, B.; Heemskerk, J.W. The glycoprotein VI-phospholipase Cg2 signaling pathway controls thrombus formation induced by collagen and tissue factor in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Rayes, J.; Watson, S.P.; Nieswandt, B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J. Clin. Investig. 2019, 129, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, M.P.; Sinha, U.; André, P.; Taylor, S.M.; Pak, Y.; Deguzman, F.R.; Nanda, N.; Pandey, A.; Stolla, M.; Bergmeier, W.; et al. PRT-060318, a novel Syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood 2011, 117, 2241–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, C.; Senba, M.; Mori, N. Anti-adult T-cell leukemia/lymphoma activity of cerdulatinib, a dual SYK/JAK kinase inhibitor. Int. J. Oncol. 2018, 53, 1681–1690. [Google Scholar] [CrossRef]
- Pollitt, A.Y.; Poulter, N.S.; Gitz, E.; Navarro-Nuñez, L.; Wang, Y.J.; Hughes, C.E.; Thomas, S.G.; Nieswandt, B.; Douglas, M.R.; Owen, D.M.; et al. Syk and Src family kinases regulate C-type lectin receptor 2 (CLEC-2)-mediated clustering of podoplanin and platelet adhesion to lymphatic endothelial cells. J. Biol. Chem. 2014, 289, 35695–35710. [Google Scholar] [CrossRef]
- Andre, P.; Morooka, T.; Sim, D.; Abe, K.; Lowell, C.; Nanda, N.; Delaney, S.; Siu, G.; Yan, Y.; Hollenbach, S.; et al. Critical role for Syk in responses to vascular injury. Blood 2011, 118, 5000–5010. [Google Scholar] [CrossRef] [Green Version]
- Onselaer, M.B.; Hardy, A.T.; Wilson, C.; Sanchez, X.; Babar, A.K.; Miller, J.L.; Watson, C.N.; Watson, S.K.; Bonna, A.; Philippou, H.; et al. Fibrin and D-dimer bind to monomeric GPVI. Blood Adv. 2017, 1, 1495–1504. [Google Scholar] [CrossRef] [Green Version]
- Farndale, R.W.; Sixma, J.J.; Barnes, M.J.; de Groot, P.G. The role of collagen in thrombosis and haemostasis. J. Thromb. Haemost. 2004, 2, 561–573. [Google Scholar] [CrossRef]
- Jung, S.M.; Takemura, Y.; Imamura, Y.; Hayashi, T.; Adachi, E.; Moroi, M. Collagen-type specificity of glycoprotein VI as a determinant of platelet adhesion. Platelets 2008, 19, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Herr, A.B.; Farndale, R.W. Structural insights into the interactions between platelet receptors and fibrillar collagen. J. Biol. Chem. 2009, 284, 19781–19785. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Moroi, M.; Soejima, K.; Nakagaki, T.; Miura, Y.; Berndt, M.C.; Gardiner, E.E.; Howes, J.M.; Pugh, N.; Bihan, D.; et al. Constitutive dimerization of glycoprotein VI (GPVI) in resting platelets is essential for binding to collagen and activation in flowing blood. J. Biol. Chem. 2012, 287, 30000–30013. [Google Scholar] [CrossRef] [PubMed]
- Poulter, N.S.; Pollitt, A.Y.; Owen, D.M.; Gardiner, E.E.; Andrews, R.K.; Shimizu, H.; Ishikawa, D.; Bihan, D.; Farndale, R.W.; Moroi, M.; et al. Clustering of glycoprotein VI (GPVI) dimers upon adhesion to collagen as a mechanism to regulate GPVI signaling in platelets. J. Thromb. Haemost. 2017, 15, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farndale, R.W.; Lisman, T.; Bihan, D.; Hamaia, S.; Smerling, C.S.; Pugh, N.; Konitsiotis, A.; Leitinger, B.; de Groot, P.G.; Jarvis, G.E.; et al. Cell-collagen interactions: The use of peptide Toolkits to investigate collagen-receptor interactions. Biochem. Soc. Trans. 2008, 36, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, J.W.M.; Sakariassen, K.S.; Zwaginga, J.J.; Brass, L.F.; Jackson, S.P.; Farndale, R.W.; Biorheology Subcommittee of the SCC of the ISTH. Collagen surfaces to measure thrombus formation under flow: Possibilities for standardization. J. Thromb. Haemost. 2011, 9, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Suzuki-Inoue, K.; Inoue, O. Novel interactions in platelet biology: CLEC-2/podoplanin and laminin/GPVI. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 191–194. [Google Scholar] [CrossRef]
- Mammadova-Bach, E.; Ollivier, V.; Loyau, S.; Schaff, M.; Dumont, B.; Favier, R.; Freyburger, G.; Latger-Cannard, V.; Nieswandt, B.; Gachet, C.; et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood 2015, 126, 683–691. [Google Scholar] [CrossRef]
- Induruwa, I.; Moroi, M.; Bonna, A.; Malcor, J.D.; Howes, J.M.; Warburton, E.A.; Farndale, R.W.; Jung, S.M. Platelet collagen receptor glycoprotein VI-dimer recognizes fibrinogen and fibrin through their D-domains, contributing to platelet adhesion and activation during thrombus formation. J. Thromb. Haemost. 2018, 16, 389–404. [Google Scholar] [CrossRef]
- Mangin, P.H.; Onselaer, M.B.; Receveur, N.; Le Lay, N.; Hardy, A.T.; Wilson, C.; Sanchez, X.; Loyau, S.; Dupuis, A.; Babar, A.K.; et al. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica 2018, 103, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Zoller, K.E.; Ginsberg, M.H.; Brugge, J.S.; Shattil, S.J. Regulation of the pp72syk protein tyrosine kinase by platelet integrin aIIbb3. EMBO J. 1997, 16, 6414–6425. [Google Scholar] [CrossRef] [PubMed]
- Obergfell, A.; Eto, K.; Mocsai, A.; Buensuceso, C.; Moores, S.L.; Brugge, J.S.; Lowell, C.A.; Shattil, S.J. Coordinate interactions of Csk, Src, and Syk kinases with aIIbb3 initiate integrin signaling to the cytoskeleton. J. Cell Biol. 2002, 157, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Van der Meijden, P.E.; Feijge, M.A.; Swieringa, F.; Gilio, K.; Nergiz-Unal, R.; Hamulyak, K.; Heemskerk, J.W. Key role of integrin αIIbβ3 signaling to Syk kinase in tissue factor-induced thrombin generation. Cell. Mol. Life Sci. 2012, 69, 3481–3492. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.G.; Morton, L.F.; Onley, D.J.; Peachey, A.R.; Messent, A.J.; Smethurst, P.A.; Tuckwell, D.S.; Farndale, R.W.; Barnes, M.J. Identification in collagen type I of an integrin alpha2 beta1-binding site containing an essential GER sequence. J. Biol. Chem. 1998, 273, 33287–33294. [Google Scholar] [CrossRef] [PubMed]
- Smethurst, P.A.; Joutsi-Korhonen, L.; O’Connor, M.N.; Wilson, E.; Jennings, N.S.; Garner, S.F.; Zhang, Y.; Knight, C.G.; Dafforn, T.R.; Buckle, A.; et al. Identification of the primary collagen-binding surface on human glycoprotein VI by site-directed mutagenesis and by a blocking phage antibody. Blood 2004, 103, 903–911. [Google Scholar] [CrossRef] [Green Version]
- De Witt, S.; Swieringa, F.; Cosemans, J.M.; Heemkerk, J.W. Thrombus formation on microspotted arrays of thrombogenic surfaces. Nat. Protocol Exchange 2014, 2014, 3309#. [Google Scholar]
- Siljander, P.; Lassila, R. Studies of adhesion-dependent platelet activation: Distinct roles for different participating receptors can be dissociated by proteolysis of collagen. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 3033–3043. [Google Scholar] [CrossRef]
- Gilio, K.; Harper, M.T.; Cosemans, J.M.E.M.; Konopatskaya, O.; Munnix, I.C.A.; Prinzen, L.; Leitges, M.; Liu, Q.; Molkentin, J.D.; Heemskerk, J.W.M.; et al. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J. Biol. Chem. 2010, 285, 23410–23419. [Google Scholar] [CrossRef]
- Van Geffen, J.P.; Brouns, S.; Batista, J.; McKinney, H.; Kempster, C.; Sivapalaratnam, S.; Baaten, C.B.; Boury, N.; Frontini, M.; Nagy, M.; et al. High-throughput elucidation of thrombus formation reveals sources of platelet function variability. Haematologica 2019, in press. [Google Scholar] [CrossRef]
- Feijge, M.A.; van Pampus, E.C.; Lacabaratz-Porret, C.; Hamulyak, K.; Lévy-Toledano, S.; Enouf, J.; Heemskerk, J.W. Inter-individual varability in Ca2+ signalling in platelets from healthy volunteers, relation with expression of endomembrane Ca2+-ATPases. Br. J. Haematol. 1998, 102, 850–859. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]
- Chin, W.W. Bootstrap cross-validation indices for PLS path model assessment. In Handbook of Partial Least Squares; Esposito Vinzi, V., Chin, W.W., Henseler, J., Wang, H., Eds.; Springer: Berlin, Germany, 2010; pp. 83–92. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microspot | Platelet Receptors | |||
| GPVI | α2β1 | GPIb | ||
| M1 | GFOGER-GPO + VWF-BP | ++ | ++ | ++ |
| M2 | CRP-XL + VWF-BP | ++ | o | ++ |
| M3 | GAOGER-GPO + VWF-BP | ++ | + | ++ |
| M4 | GFOGER-GPP + VWF-BP | (o)* | ++ | ++ |
| M5 | VWF-BP | o | o | ++ |
| M6 | collagen-H (Horm type) | ++ | ++ | ++ |
| M7 | collagen-I (human) | n.a. | n.a. | ++ |
| M8 | monomeric collagen-I (human) | n.a. | n.a. | ++ |
| M9 | collagen-III (human) | n.a. | n.a. | ++ |
| Parameter | range | scaling | ||
| Bright-Field Images | ||||
| P1 | platelet deposition (% SAC) | 0–51.52 | 0–10 | |
| P2 | platelet aggregate coverage (% SAC) | 0–21.09 | 0–10 | |
| P3 | thrombus morphological score | 0–4.10 | 0–10 | |
| P4 | thrombus multilayer score | 0–2.60 | 0–10 | |
| P5 | thrombus contraction score | 0–2.94 | 0–10 | |
| Fluorescence Images | ||||
| P6 | PS exposure (% SAC) | 0–13.91 | 0–10 | |
| P7 | CD62P expression (% SAC) | 0–46.71 | 0–10 | |
| P8 | fibrinogen binding (% SAC) | 0–28.33 | 0–10 | |
| Microspot | GPVI Dependency | Correctly Predicted | ||
|---|---|---|---|---|
| Range | Ctrl | Syk-IN | ||
| M1 | positive | 0.41–1.06 | 5/6 | 6/6 |
| M2 | positive | 0.27–0.76 | 4/5 | 5/5 |
| M3 | positive | 0.86–1.07 | 5/5 | 5/5 |
| M4 | negative | 0.57–0.97 | 0/6 | 6/6 |
| M5 | negative | 0.21–0.34 | 5/5 | 5/5 |
| M6 | positive | 0.68–1.11 | 7/7 | 6/7 |
| M7 | mixed | 0.44–0.85 | 5/7 | 7/7 |
| M8 | negative | 0.13–0.41 | 0/5 | 5/5 |
| M9 | mixed | 0.49–0.67 | 3/5 | 5/5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jooss, N.J.; De Simone, I.; Provenzale, I.; Fernández, D.I.; Brouns, S.L.N.; Farndale, R.W.; Henskens, Y.M.C.; Kuijpers, M.J.E.; ten Cate, H.; van der Meijden, P.E.J.; et al. Role of Platelet Glycoprotein VI and Tyrosine Kinase Syk in Thrombus Formation on Collagen-Like Surfaces. Int. J. Mol. Sci. 2019, 20, 2788. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112788
Jooss NJ, De Simone I, Provenzale I, Fernández DI, Brouns SLN, Farndale RW, Henskens YMC, Kuijpers MJE, ten Cate H, van der Meijden PEJ, et al. Role of Platelet Glycoprotein VI and Tyrosine Kinase Syk in Thrombus Formation on Collagen-Like Surfaces. International Journal of Molecular Sciences. 2019; 20(11):2788. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112788
Chicago/Turabian StyleJooss, Natalie J., Ilaria De Simone, Isabella Provenzale, Delia I. Fernández, Sanne L.N. Brouns, Richard W. Farndale, Yvonne M.C. Henskens, Marijke J.E. Kuijpers, Hugo ten Cate, Paola E.J. van der Meijden, and et al. 2019. "Role of Platelet Glycoprotein VI and Tyrosine Kinase Syk in Thrombus Formation on Collagen-Like Surfaces" International Journal of Molecular Sciences 20, no. 11: 2788. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112788