Leloir Glycosyltransferases in Applied Biocatalysis: A Multidisciplinary Approach

 , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Glycosyltransferases in Nature
2.1. Distinguishing Glycosyl Transferases from Glycoside Hydrolases
2.2. Recombinant Expression of Glycosyl Transferases
3. Application of Glycosyl Transferases in Organic Synthesis
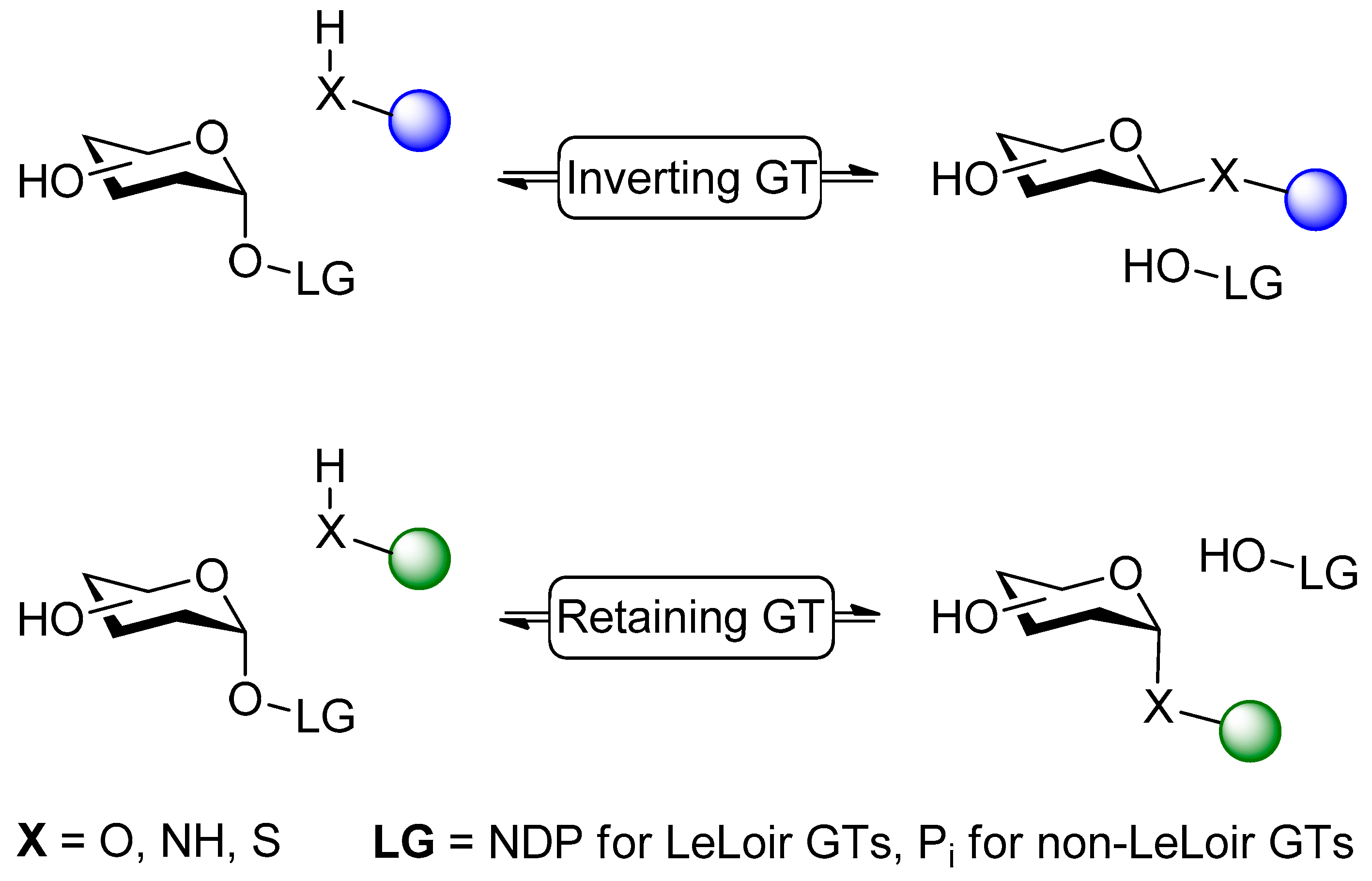
3.1. Catalytic Reversibility of Glycosyltransferases
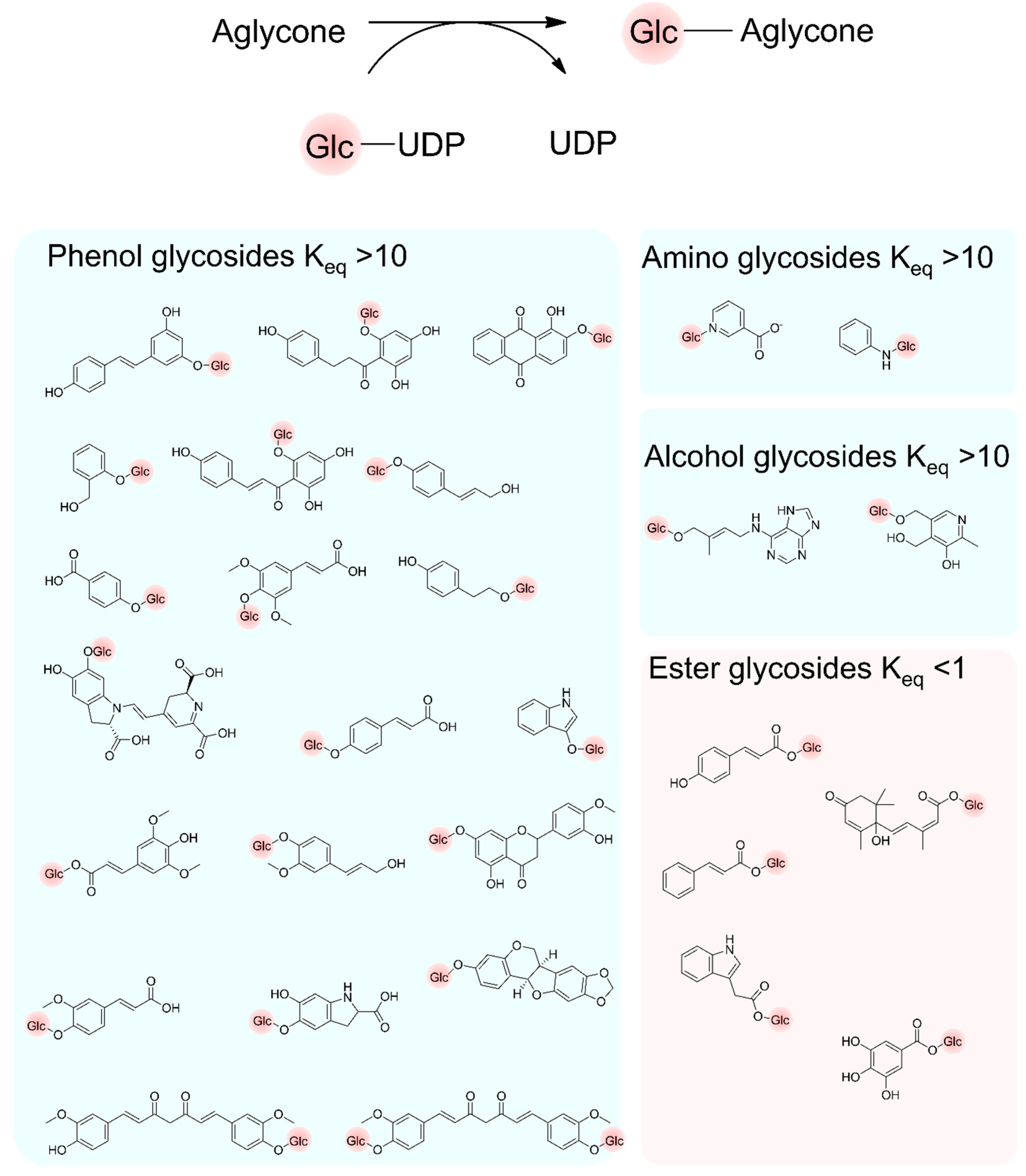
3.2. Sugar Donors and Acceptors and Their Glycosylation Efficiency
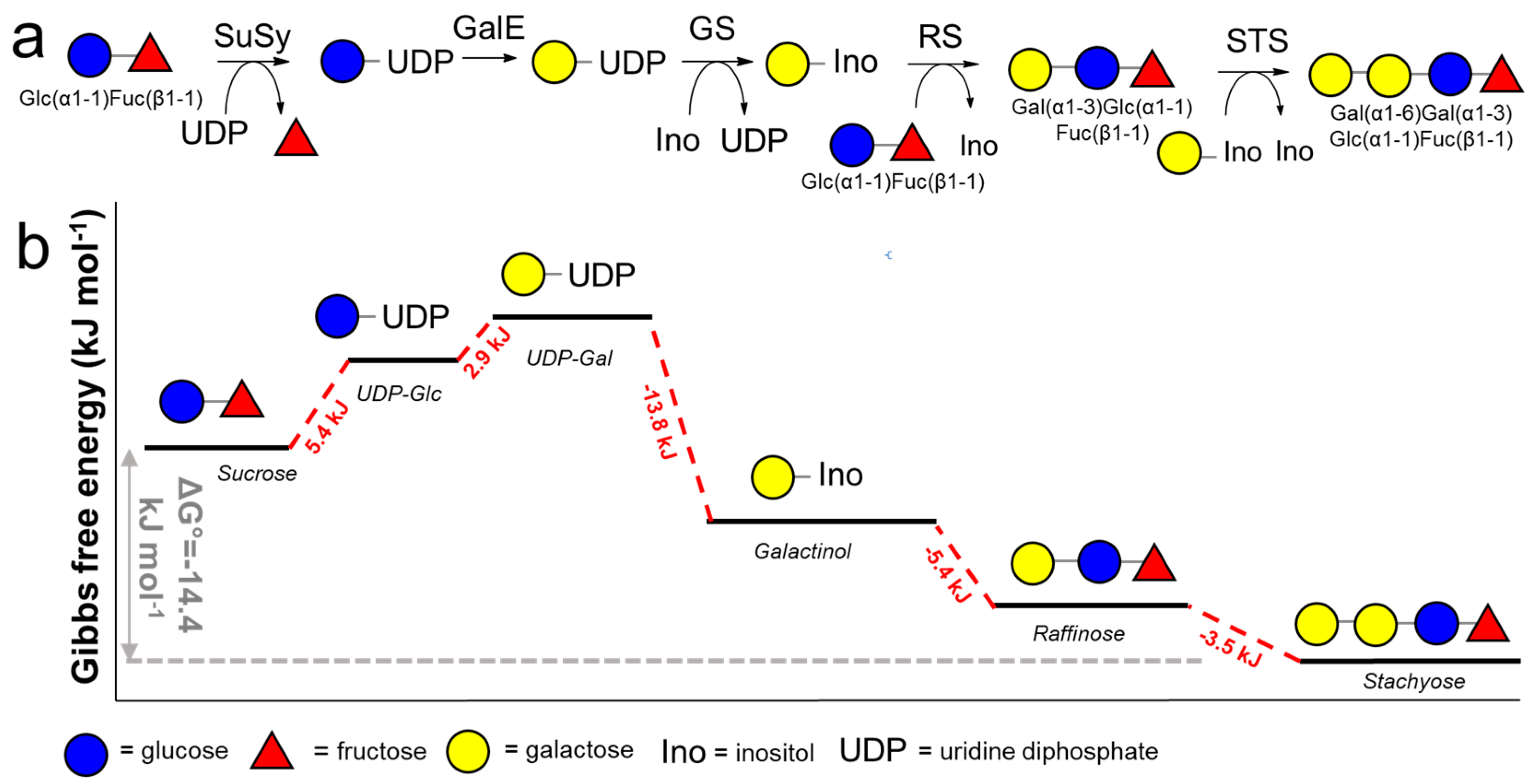
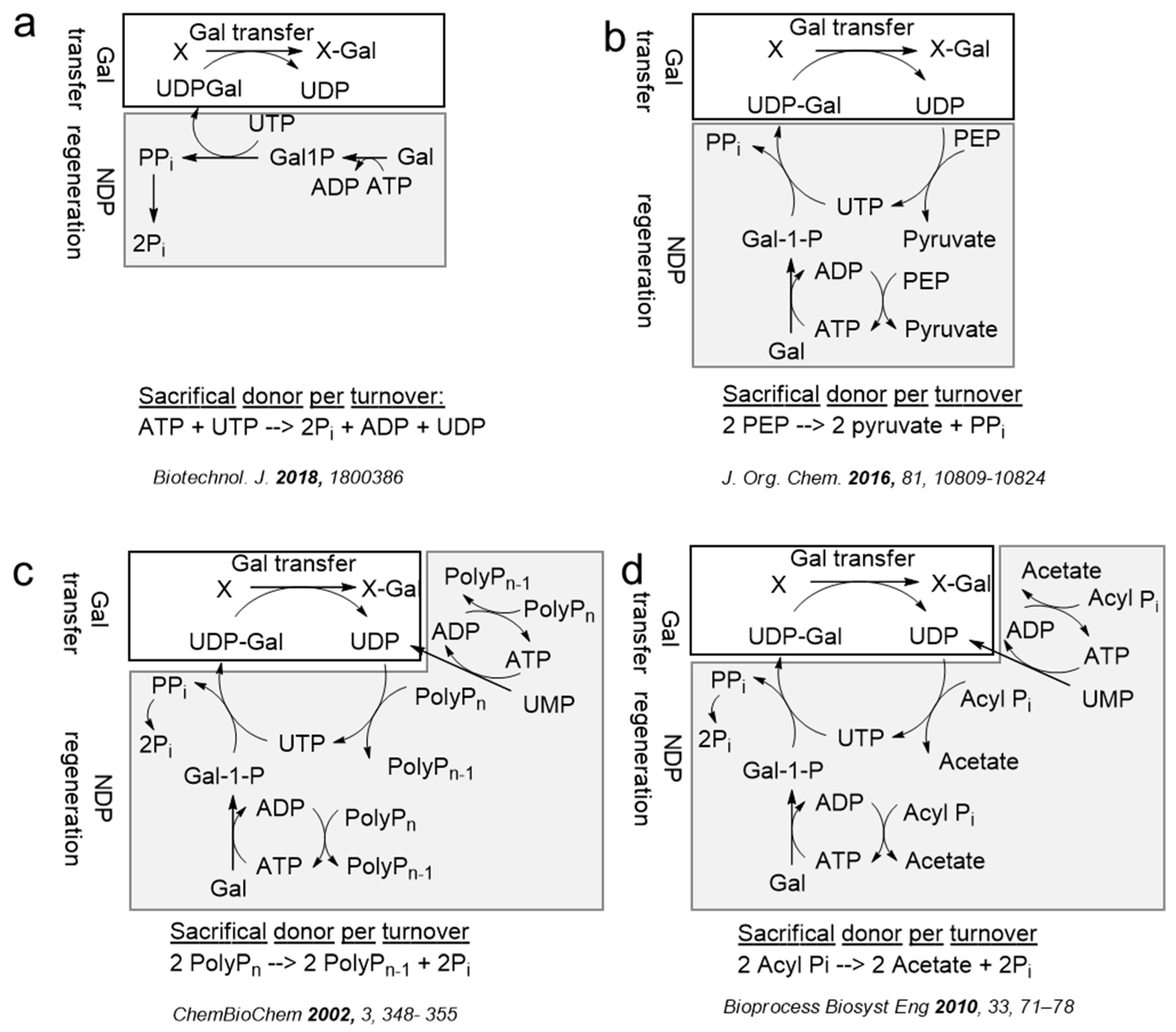
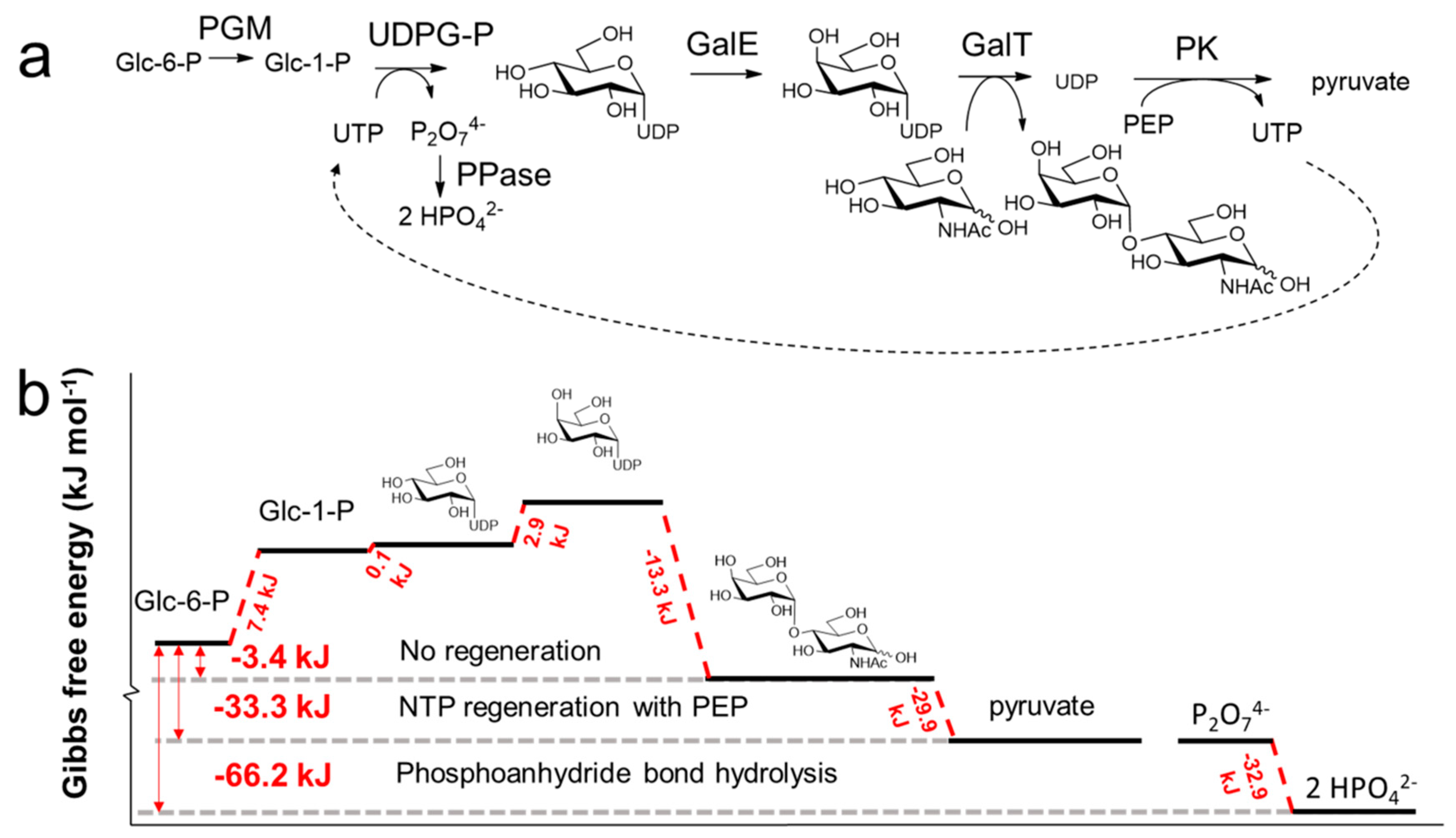
3.3. NTP Regeneration for NDP-Sugar Donor Production
3.4. Chemoenzymatic NTP Regeneration Cascades
3.5. One-Pot Multi Enzyme Cascades
4. Reactor Engineering for (Non)-LeLoir Glycosyltransferases
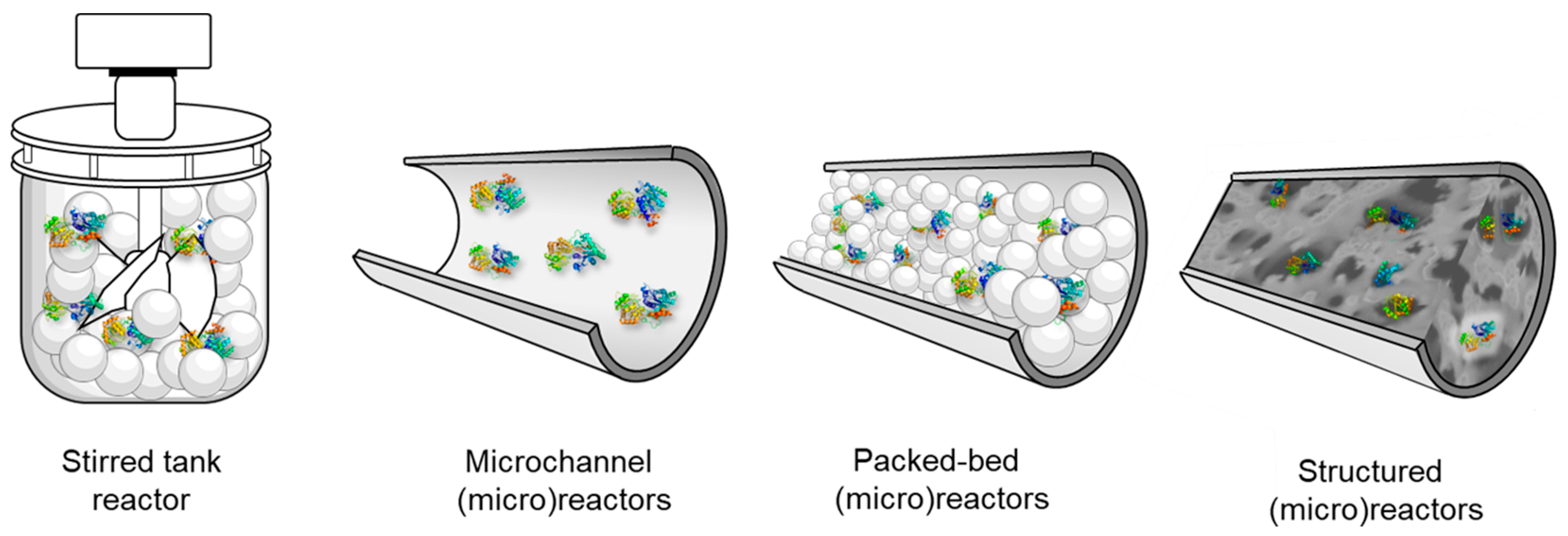
Reactor Design for Glycosyltransferases
5. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Fischer, E. Ueber die configuration des traubenzuckers und seiner isomeren. Ber. Dtsch. Chem. Ges. 1891, 24, 1836–1845. [Google Scholar] [CrossRef]
- Fischer, E. Ueber die configuration des traubenzuckers und seiner isomeren. II. Ber. Dtsch. Chem. Ges. 1891, 24, 2683–2687. [Google Scholar] [CrossRef]
- Wöhler, F.; Liebig, J. Ueber die bildung des bittermandelöls. Ann. Pharm. 1837, 22, 1–24. [Google Scholar]
- DiCosimo, R.; McAuliffe, J.; Poulose, A.J.; Bohlmann, G. Industrial use of immobilized enzymes. Chem. Soc. Rev. 2013, 42, 6437–6474. [Google Scholar] [CrossRef] [PubMed]
- Laine, R.A. A calculation of all possible oligosaccharide isomers both branched and linear yields 1.05 × 1012 structures for a reducing hexasaccharide: The isomer barrier to development of single-method saccharide sequencing or synthesis systems. Glycobiology 1994, 4, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Aebi, M.; Packer, N.H.; Seeberger, P.H.; Esko, J.D.; Stanley, P.; Hart, G.; Darvill, A.; Kinoshita, T.; et al. Symbol nomenclature for graphical representations of glycans. Glycobiology 2015, 25, 1323–1324. [Google Scholar] [CrossRef]
- Puri, M.; Kaur, A.; Singh, R.S.; Kanwar, J.R. Immobilized Enzyme Technology for Debittering Citrus Fruit Juices; Transworld Research Network: Kerala, India, 2008; pp. 91–103. [Google Scholar]
- Vila Real, H.J.; Alfaia, A.J.; Calado, A.R.T.; Ribeiro, M.H.L. High pressure-temperature effects on enzymatic activity: Naringin bioconversion. Food Chem. 2007, 102, 565–570. [Google Scholar] [CrossRef]
- Puri, M.; Banerjee, U.C. Production, purification, and characterization of the debittering enzyme naringinase. Biotechnol. Adv. 2000, 18, 207–217. [Google Scholar] [CrossRef]
- Kubota, M. New function and property of trehalose. New Food Ind. 2005, 47, 17–29. [Google Scholar]
- Walmagh, M.; Zhao, R.; Desmet, T. Trehalose analogues: Latest insights in properties and biocatalytic production. Int. J. Mol. Sci. 2015, 16, 13729. [Google Scholar] [CrossRef]
- Szaniawski, A.R.; Spencer, H.G. Effects of immobilized pectinase on the microfiltration of dilute pectin solutions by macroporous titania membranes: Resistance model interpretation. J. Membr. Sci. 1997, 127, 69–76. [Google Scholar] [CrossRef]
- Alkorta, I.; Garbisu, C.; Llama, M.J.; Serra, J.L. Industrial applications of pectic enzymes: A review. Process Biochem. 1998, 33, 21–28. [Google Scholar] [CrossRef]
- Lozano, P.; Manjón, A.; Iborra, J.; Cánovas, M.; Romojaro, F. Kinetic and operational study of a cross-flow reactor with immobilized pectolytic enzymes. Enzyme Microb. Technol. 1990, 12, 499–505. [Google Scholar] [CrossRef]
- Lozano, P.; Manjón, A.; Romojaro, F.; Canovas, M.; Iborra, J.L. A cross-flow reactor with immobilized pectolytic enzymes for juice clarification. Biotechnol. Lett. 1987, 9, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Garbisu, C.; Llama, M.J.; Serra, J.L. Viscosity decrease of pectin and fruit juices catalyzed by pectin lyase from Penicillium italicum in batch and continuous-flow membrane reactors. Biotechnol. Tech. 1995, 9, 95–100. [Google Scholar] [CrossRef]
- Lisboa, M.P.; Khan, N.; Martin, C.; Xu, F.-F.; Reppe, K.; Geissner, A.; Govindan, S.; Witzenrath, M.; Pereira, C.L.; Seeberger, P.H. Semisynthetic glycoconjugate vaccine candidate against Streptococcus pneumoniae serotype 5. Proc. Natl. Acad. Sci. USA 2017, 114, 11063–11068. [Google Scholar] [CrossRef]
- Emmadi, M.; Khan, N.; Lykke, L.; Reppe, K.G.; Parameswarappa, S.; Lisboa, M.P.; Wienhold, S.-M.; Witzenrath, M.; Pereira, C.L.; Seeberger, P.H. A Streptococcus pneumoniae type 2 oligosaccharide glycoconjugate elicits opsonic antibodies and is protective in an animal model of invasive pneumococcal disease. J. Am. Chem. Soc. 2017, 139, 14783–14791. [Google Scholar] [CrossRef]
- Cavallari, M.; Stallforth, P.; Kalinichenko, A.; Rathwell, D.C.K.; Gronewold, T.M.A.; Adibekian, A.; Mori, L.; Landmann, R.; Seeberger, P.H.; De Libero, G. A semisynthetic carbohydrate-lipid vaccine that protects against S. pneumoniae in mice. Nat. Chem. Biol. 2014, 10, 950. [Google Scholar] [CrossRef]
- Oldrini, D.; Fiebig, T.; Romano, M.R.; Proietti, D.; Berger, M.; Tontini, M.; De Ricco, R.; Santini, L.; Morelli, L.; Lay, L.; et al. Combined chemical synthesis and tailored enzymatic elongation provide fully synthetic and conjugation-ready Neisseria meningitidis serogroup x vaccine antigens. ACS Chem. Biol. 2018, 13, 984–994. [Google Scholar] [CrossRef]
- Marciani, D.J.; Press, J.B.; Reynolds, R.C.; Pathak, A.K.; Pathak, V.; Gundy, L.E.; Farmer, J.T.; Koratich, M.S.; May, R.D. Development of semisynthetic triterpenoid saponin derivatives with immune stimulating activity. Vaccine 2000, 18, 3141–3151. [Google Scholar] [CrossRef]
- Fiebig, T.; Romano, M.R.; Oldrini, D.; Adamo, R.; Tontini, M.; Brogioni, B.; Santini, L.; Berger, M.; Costantino, P.; Berti, F.; et al. An efficient cell free enzyme-based total synthesis of a meningococcal vaccine candidate. NPJ Vaccines 2016, 1, 16017. [Google Scholar] [CrossRef] [PubMed]
- Moremen, K.W.; Ramiah, A.; Stuart, M.; Steel, J.; Meng, L.; Forouhar, F.; Moniz, H.A.; Gahlay, G.; Gao, Z.; Chapla, D.; et al. Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol. 2017, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Edmunds, G.; Gibbons, C.; Zhang, J.; Gadi, M.R.; Zhu, H.; Fang, J.; Liu, X.; Kong, Y.; Wang, P.G. Toward automated enzymatic synthesis of oligosaccharides. Chem. Rev. 2018, 118, 8151–8187. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn, F.; Maertens, J.; Beauprez, J.; Soetaert, W.; De Mey, M. Biotechnological advances in UDP-sugar based glycosylation of small molecules. Biotechnol. Adv. 2015, 33, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Ann. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Gloster, T.M. Advances in understanding glycosyltransferases from a structural perspective. Curr. Opin. Struct. Biol. 2014, 28, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, C.; Trent, M.S. Biosynthesis and export of bacterial lipopolysaccharides. Ann. Rev. Biochem. 2014, 83, 99–128. [Google Scholar] [CrossRef]
- Typas, A.; Banzhaf, M.; Gross, C.A.; Vollmer, W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 2011, 10, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The carbohydrate-active enzymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Campbell, J.A.; Davies, G.J.; Bulone, V.; Henrissat, B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 1997, 326 Pt 3, 929–939. [Google Scholar] [CrossRef]
- Sinnott, M. Carbohydrate Chemistry and Biochemistry: Structure and Mechanism; Royal Society of Chemistry: Cambridge, UK, 2013; p. 1, online resource. [Google Scholar]
- Tvaroška, I. Atomistic insight into the catalytic mechanism of glycosyltransferases by combined quantum mechanics/molecular mechanics (QM/MM) methods. Carbohydr. Res. 2015, 403, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Welzel, P. Syntheses around the transglycosylation step in peptidoglycan biosynthesis. Chem. Rev. 2005, 105, 4610–4660. [Google Scholar] [CrossRef]
- Lovering, A.L.; de Castro, L.H.; Lim, D.; Strynadka, N.C.J. Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science 2007, 315, 1402–1405. [Google Scholar] [CrossRef] [PubMed]
- Timm, M.; Görl, J.; Kraus, M.; Kralj, S.; Hellmuth, H.; Beine, R.; Buchholz, K.; Dijkhuizen, L.; Seibel, J. An unconventional glycosyl transfer reaction: Glucansucrase GTFA functions as an allosyltransferase enzyme. ChemBioChem 2013, 14, 2423–2426. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Hubbard, C.; Purushotham, P.; Zimmer, J. Insights into the structure and function of membrane-integrated processive glycosyltransferases. Curr. Opin. Struct. Biol. 2015, 34, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton, C.; Snajdrová, L.; Jeanneau, C.; Koca, J.; Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 2006, 16, 29R–37R. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, F.; Yang, T.; Ding, L.; Zhou, M.; Li, J.; Haslam, S.M.; Dell, A.; Erlandsen, H.; Wu, H. The highly conserved domain of unknown function 1792 has a distinct glycosyltransferase fold. Nat. Commun. 2014, 5, 4339. [Google Scholar] [CrossRef]
- Kattke, M.D.; Gosschalk, J.E.; Martinez, O.E.; Kumar, G.; Gale, R.T.; Cascio, D.; Sawaya, M.R.; Philips, M.; Brown, E.D.; Clubb, R.T. Structure and mechanism of TagA, a novel membrane-associated glycosyltransferase that produces wall teichoic acids in pathogenic bacteria. PLoS Pathog. 2019, 15, e1007723. [Google Scholar] [CrossRef] [PubMed]
- Ardèvol, A.; Rovira, C. Reaction mechanisms in carbohydrate-active enzymes: Glycoside hydrolases and glycosyltransferases. Insights from ab initio quantum mechanics/molecular mechanics dynamic simulations. J. Am. Chem. Soc. 2015, 137, 7528–7547. [Google Scholar] [CrossRef]
- Gutmann, A.; Lepak, A.; Diricks, M.; Desmet, T.; Nidetzky, B. Glycosyltransferase cascades for natural product glycosylation: Use of plant instead of bacterial sucrose synthases improves the UDP-glucose recycling from sucrose and UDP. Biotechnol. J. 2017, 12, 1600557. [Google Scholar] [CrossRef]
- Ryu, S.-I.; Kim, J.-E.; Kim, E.-J.; Chung, S.-K.; Lee, S.-B. Catalytic reversibility of Pyrococcus horikoshii trehalose synthase: Efficient synthesis of several nucleoside diphosphate glucoses with enzyme recycling. Proc. Biochem. 2011, 46, 128–134. [Google Scholar] [CrossRef]
- Nidetzky, B.; Gutmann, A.; Zhong, C. Leloir glycosyltransferases as biocatalysts for chemical production. ACS Catal. 2018, 8, 6283–6300. [Google Scholar] [CrossRef]
- Chang, A.; Singh, S.; Phillips, G.N.; Thorson, J.S. Glycosyltransferase structural biology and its role in the design of catalysts for glycosylation. Curr. Opin. Biotechnol. 2011, 22, 800–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton, C.; Fournel-Gigleux, S.; Palcic, M.M. Recent structures, evolution and mechanisms of glycosyltransferases. Curr. Opin. Struct. Biol. 2012, 22, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Blanco Capurro, J.I.; Hopkins, C.W.; Pierdominici Sottile, G.; González Lebrero, M.C.; Roitberg, A.E.; Marti, M.A. Theoretical insights into the reaction and inhibition mechanism of metal-independent retaining glycosyltransferase responsible for mycothiol biosynthesis. J. Phys. Chem. B 2017, 121, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Albesa-Jové, D.; Sainz-Polo, M.Á.; Marina, A.; Guerin, M.E. Structural snapshots of α-1,3-galactosyltransferase with native substrates: Insight into the catalytic mechanism of retaining glycosyltransferases. Angew. Chem. Int. Ed. 2017, 129, 15049–15053. [Google Scholar] [CrossRef]
- Charnock, S.J.; Davies, G.J. Structure of the nucleotide-diphospho-sugar transferase, spsa from Bacillus subtilis, in native and nucleotide-complexed forms. Biochemistry 1999, 38, 6380–6385. [Google Scholar] [CrossRef]
- Huber, R.E.; Gaunt, M.T.; Hurlburt, K.L. Binding and reactivity at the “glucose” site of galactosyl-β-galactosidase (Escherichia coli). Arch. Biochem. Biophys. 1984, 234, 151–160. [Google Scholar] [CrossRef]
- Ooi, Y.; Mitsuo, N.; Satoh, T. Enzymic synthesis of glycosides of racemic alcohols using beta-galactosidase and separation of the diastereomers by high-performance liquid chromatography using a conventional column. Chem. Pharm. Bull. 1985, 33, 5547–5550. [Google Scholar] [CrossRef]
- Okuyama, M.; Mori, H.; Watanabe, K.; Kimura, A.; Chiba, S. A-glucosidase mutant catalyzes “α-glycosynthase”-type reaction. Biosci. Biotechnol. Biochem. 2002, 66, 928–933. [Google Scholar] [CrossRef]
- Jahn, M.; Marles, J.; Warren, R.A.J.; Withers, S.G. Thioglycoligases: Mutant glycosidases for thioglycoside synthesis. Angew. Chem. Int. Ed. 2003, 42, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.; Peiru, S.; Eberhardt, F.; Vetcher, L.; Cabrera, R.; Menzella, H.G. Enzymatic hydrolysis of steryl glucosides, major contaminants of vegetable oil-derived biodiesel. Appl. Microbiol. Biotechnol. 2014, 98, 4033–4040. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Su, H.; Mi, S.; Han, Y. A multifunctional thermophilic glycoside hydrolase from Caldicellulosiruptor owensensis with potential applications in production of biofuels and biochemicals. Biotechnol. Biofuels 2016, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Ezeilo, U.R.; Zakaria, I.I.; Huyop, F.; Wahab, R.A. Enzymatic breakdown of lignocellulosic biomass: The role of glycosyl hydrolases and lytic polysaccharide monooxygenases. Biotechnol. Biotechnol. Equip. 2017, 31, 647–662. [Google Scholar] [CrossRef]
- Michlmayr, H.; Varga, E.; Malachova, A.; Nguyen, N.T.; Lorenz, C.; Haltrich, D.; Berthiller, F.; Adam, G. A versatile family 3 glycoside hydrolase from Bifidobacterium adolescentis hydrolyzes β-glucosides of the Fusarium mycotoxins deoxynivalenol, nivalenol, and HT-2 toxin in cereal matrices. Appl. Environ. Microbiol. 2015, 81, 4885–4893. [Google Scholar] [CrossRef]
- Mitsuo, N.; Takeichi, H.; Satoh, T. Synthesis of beta-alkyl glucosides by enzymic transglucosylation. Chem. Pharm. Bull. 1984, 32, 1183–1187. [Google Scholar] [CrossRef]
- Drueckhammer, D.G.; Wong, C.H. Chemoenzymic syntheses of fluoro sugar phosphates and analogs. J. Org. Chem. 1985, 50, 5912–5913. [Google Scholar] [CrossRef]
- Gold, A.M.; Osber, M.P. A-d-glucopyranosyl fluoride: A substrate of sucrose phosphorylase. Biochem. Biophys. Res. Commun. 1971, 42, 469–474. [Google Scholar] [CrossRef]
- Williams, S.J.; Withers, S.G. Glycosyl fluorides in enzymatic reactions. Carbohydr. Res. 2000, 327, 27–46. [Google Scholar] [CrossRef]
- Tolborg, J.F.; Petersen, L.; Jensen, K.J.; Mayer, C.; Jakeman, D.L.; Warren, R.A.J.; Withers, S.G. Solid-phase oligosaccharide and glycopeptide synthesis using glycosynthases. J. Org. Chem. 2002, 67, 4143–4149. [Google Scholar] [CrossRef]
- Fialová, P.; Carmona, A.T.; Robina, I.; Ettrich, R.; Sedmera, P.; Přikrylová, V.; Petrásková-Hušáková, L.; Křen, V. Glycosyl azide—A novel substrate for enzymatic transglycosylations. Tetrahedron Lett. 2005, 46, 8715–8718. [Google Scholar] [CrossRef]
- Strahsburger, E.; de Lacey, A.M.L.; Marotti, I.; DiGioia, D.; Biavati, B.; Dinelli, G. In vivo assay to identify bacteria with β-glucosidase activity. Electr. J. Biotechnol. 2017, 30, 83–87. [Google Scholar] [CrossRef]
- Yasukochi, T.; Fukase, K.; Kusumoto, S. 3-nitro-2-pyridyl glycoside as donor for chemical glycosylation and its application to chemoenzymatic synthesis of oligosaccharide. Tetrahedron Lett. 1999, 40, 6591–6593. [Google Scholar] [CrossRef]
- Chiffoleau-Giraud, V.; Spangenberg, P.; Rabiller, C. Β-galactosidase transferase activity in ice and use of vinyl-β-d-galactoside as donor. Tetrahedron Asymmetry 1997, 8, 2017–2023. [Google Scholar] [CrossRef]
- Vic, G.; Crout, D.H.G. Synthesis of allyl and benzyl β-d-glucopyranosides, and allyl β-d-galactopyranoside from d-glucose or d-galactose and the corresponding alcohol using almond β-d-glucosidase. Carbohydr. Res. 1995, 279, 315–319. [Google Scholar] [CrossRef]
- Hansson, T.; Andersson, M.; Wehtje, E.; Adlercreutz, P. Influence of water activity on the competition between β-glycosidase-catalysed transglycosylation and hydrolysis in aqueous hexanol. Enzyme Microb. Technol. 2001, 29, 527–534. [Google Scholar] [CrossRef]
- Gavlighi, H.A.; Meyer, A.S.; Mikkelsen, J.D. Enhanced enzymatic cellulose degradation by cellobiohydrolases via product removal. Biotechnol. Lett. 2013, 35, 205–212. [Google Scholar] [CrossRef]
- Cote, J.M.; Taylor, E.A. The glycosyltransferases of lps core: A review of four heptosyltransferase enzymes in context. Int. J. Mol. Sci. 2017, 18, 2256. [Google Scholar] [CrossRef]
- Moréra, S.; Imberty, A.; Aschke-Sonnenborn, U.; Rüger, W.; Freemont, P.S. T4 phage β-glucosyltransferase: Substrate binding and proposed catalytic mechanism. J. Mol. Biol. 1999, 292, 717–730. [Google Scholar] [CrossRef]
- Czabany, T.; Schmölzer, K.; Luley-Goedl, C.; Ribitsch, D.; Nidetzky, B. All-in-one assay for β-d-galactoside sialyltransferases: Quantification of productive turnover, error hydrolysis, and site selectivity. Anal. Biochem. 2015, 483, 47–53. [Google Scholar] [CrossRef]
- Ding, L.; Zhao, C.; Qu, J.; Li, Y.; Sugiarto, G.; Yu, H.; Wang, J.; Chen, X. A Photobacterium sp. α2–6-sialyltransferase (Psp2,6ST) mutant with an increased expression level and improved activities in sialylating Tn antigens. Carbohydr. Res. 2015, 408, 127–133. [Google Scholar] [CrossRef]
- Schmölzer, K.; Luley-Goedl, C.; Czabany, T.; Ribitsch, D.; Schwab, H.; Weber, H.; Nidetzky, B. Mechanistic study of CMP-Neu5Ac hydrolysis by α2,3-sialyltransferase from Pasteurella dagmatis. FEBS Lett. 2014, 588, 2978–2984. [Google Scholar] [CrossRef] [PubMed]
- Sugiarto, G.; Lau, K.; Qu, J.; Li, Y.; Lim, S.; Mu, S.; Ames, J.B.; Fisher, A.J.; Chen, X. A sialyltransferase mutant with decreased donor hydrolysis and reduced sialidase activities for directly sialylating Lewisx. ACS Chem. Biol. 2012, 7, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Schmölzer, K.; Eibinger, M.; Nidetzky, B. Active-site his85 of Pasteurella dagmatis sialyltransferase facilitates productive sialyl transfer and so prevents futile hydrolysis of CMP-Neu5Ac. ChemBioChem 2017, 18, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wu, Y.; Yu, H.; Shah, I.M.; Li, Y.; Zeng, J.; Liu, B.; Mills, D.A.; Chen, X. One-pot multienzyme (OPME) synthesis of human blood group H antigens and a human milk oligosaccharide (HMOS) with highly active Thermosynechococcus elongatus α1–2-fucosyltransferase. Chem. Commun. 2016, 52, 3899–3902. [Google Scholar] [CrossRef]
- Koszagova, R.; Krajcovic, T.; Palencarova-Talafova, K.; Patoprsty, V.; Vikartovska, A.; Pospiskova, K.; Safarik, I.; Nahalka, J. Magnetization of active inclusion bodies: Comparison with centrifugation in repetitive biotransformations. Microb. Cell Factories 2018, 17, 139. [Google Scholar] [CrossRef]
- Mestrom, L.; Marsden, S.R.; Dieters, M.; Achterberg, P.; Stolk, L.; Bento, I.; Hanefeld, U.; Hagedoorn, P.-L. Artificial fusion of mCherry enhanced solubility and stability of trehalose transferase. Appl. Environ. Microbiol. 2019, 85, e03084-18. [Google Scholar]
- Schmölzer, K.; Gutmann, A.; Diricks, M.; Desmet, T.; Nidetzky, B. Sucrose synthase: A unique glycosyltransferase for biocatalytic glycosylation process development. Biotechnol. Adv. 2016, 34, 88–111. [Google Scholar] [CrossRef]
- Bungaruang, L.; Gutmann, A.; Nidetzky, B. Leloir glycosyltransferases and natural product glycosylation: Biocatalytic synthesis of the C-glucoside nothofagin, a major antioxidant of redbush herbal tea. Adv. Synth. Catal. 2013, 355, 2757–2763. [Google Scholar] [CrossRef]
- Schmölzer, K.; Lemmerer, M.; Nidetzky, B. Glycosyltransferase cascades made fit for chemical production: Integrated biocatalytic process for the natural polyphenol C-glucoside nothofagin. Biotechnol. Bioeng. 2018, 115, 545–556. [Google Scholar] [CrossRef]
- Wang, Q.M.; Peery, R.B.; Johnson, R.B.; Alborn, W.E.; Yeh, W.-K.; Skatrud, P.L. Identification and characterization of a monofunctional glycosyltransferase from Staphylococcus aureus. J. Bacteriol. 2001, 183, 4779–4785. [Google Scholar] [CrossRef]
- Chen, L.; Sun, P.; Li, Y.; Yan, M.; Xu, L.; Chen, K.; Ouyang, P. A fusion protein strategy for soluble expression of stevia glycosyltransferase UGT76G1 in Escherichia coli. 3 Biotech 2017, 7, 356. [Google Scholar] [CrossRef]
- Welner, D.H.; Shin, D.; Tomaleri, G.P.; DeGiovanni, A.M.; Tsai, A.Y.-L.; Tran, H.M.; Hansen, S.F.; Green, D.T.; Scheller, H.V.; Adams, P.D. Plant cell wall glycosyltransferases: High-throughput recombinant expression screening and general requirements for these challenging enzymes. PLoS ONE 2017, 12, e0177591. [Google Scholar] [CrossRef]
- Ortiz-Soto, M.E.; Seibel, J. Expression of functional human sialyltransferases ST3Gal1 and ST6Gal1 in Escherichia coli. PLoS ONE 2016, 11, e0155410. [Google Scholar] [CrossRef] [PubMed]
- Plante, O.J.; Palmacci, E.R.; Seeberger, P.H. Automated solid-phase synthesis of oligosaccharides. Science 2001, 291, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Pistorio, S.G.; Nigudkar, S.S.; Stine, K.J.; Demchenko, A.V. HPLC-assisted automated oligosaccharide synthesis: Implementation of the autosampler as a mode of the reagent delivery. J. Org. Chem. 2016, 81, 8796–8805. [Google Scholar] [CrossRef]
- Nokami, T.; Hayashi, R.; Saigusa, Y.; Shimizu, A.; Liu, C.-Y.; Mong, K.-K.T.; Yoshida, J.-I. Automated solution-phase synthesis of oligosaccharides via iterative electrochemical assembly of thioglycosides. Org. Lett. 2013, 15, 4520–4523. [Google Scholar] [CrossRef]
- Huang, T.-Y.; Zulueta, M.M.L.; Hung, S.-C. Regioselective one-pot protection, protection–glycosylation and protection–glycosylation–glycosylation of carbohydrates: A case study with d-glucose. Org. Biomol. Chem. 2014, 12, 376–382. [Google Scholar] [CrossRef]
- Wang, C.-C.; Lee, J.-C.; Luo, S.-Y.; Kulkarni, S.S.; Huang, Y.-W.; Lee, C.-C.; Chang, K.-L.; Hung, S.-C. Regioselective one-pot protection of carbohydrates. Nature 2007, 446, 896. [Google Scholar] [CrossRef]
- Walvoort, M.T.C.; Volbeda, A.G.; Reintjens, N.R.M.; van den Elst, H.; Plante, O.J.; Overkleeft, H.S.; van der Marel, G.A.; Codée, J.D.C. Automated solid-phase synthesis of hyaluronan oligosaccharides. Org. Lett. 2012, 14, 3776–3779. [Google Scholar] [CrossRef]
- Geert Volbeda, A.; van Mechelen, J.; Meeuwenoord, N.; Overkleeft, H.S.; van der Marel, G.A.; Codée, J.D.C. Cyanopivaloyl ester in the automated solid-phase synthesis of oligorhamnans. J. Org. Chem. 2017, 82, 12992–13002. [Google Scholar] [CrossRef]
- Zhou, J.; Lv, S.; Zhang, D.; Xia, F.; Hu, W. Deactivating influence of 3-O-glycosyl substituent on anomeric reactivity of thiomannoside observed in oligomannoside synthesis. J. Org. Chem. 2017, 82, 2599–2621. [Google Scholar] [CrossRef]
- Kaeothip, S.; Demchenko, A.V. On orthogonal and selective activation of glycosyl thioimidates and thioglycosides: Application to oligosaccharide assembly. J. Org. Chem. 2011, 76, 7388–7398. [Google Scholar] [CrossRef]
- Kanie, O.; Ito, Y.; Ogawa, T. Orthogonal glycosylation strategy in oligosaccharide synthesis. J. Am. Chem. Soc. 1994, 116, 12073–12074. [Google Scholar] [CrossRef]
- Tang, S.-L.; Linz, L.B.; Bonning, B.C.; Pohl, N.L.B. Automated solution-phase synthesis of insect glycans to probe the binding affinity of pea enation mosaic virus. J. Org. Chem. 2015, 80, 10482–10489. [Google Scholar] [CrossRef]
- Senf, D.; Ruprecht, C.; de Kruijff, G.H.M.; Simonetti, S.O.; Schuhmacher, F.; Seeberger, P.H.; Pfrengle, F. Active site mapping of xylan-deconstructing enzymes with arabinoxylan oligosaccharides produced by automated glycan assembly. Chem. Eur. J. 2017, 23, 3197–3205. [Google Scholar] [CrossRef]
- Naresh, K.; Schumacher, F.; Hahm, H.S.; Seeberger, P.H. Pushing the limits of automated glycan assembly: Synthesis of a 50mer polymannoside. Chem. Commun. 2017, 53, 9085–9088. [Google Scholar] [CrossRef]
- Weishaupt, M.W.; Hahm, H.S.; Geissner, A.; Seeberger, P.H. Automated glycan assembly of branched β-(1,3)-glucans to identify antibody epitopes. Chem. Commun. 2017, 53, 3591–3594. [Google Scholar] [CrossRef]
- Hahm, H.S.; Hurevich, M.; Seeberger, P.H. Automated assembly of oligosaccharides containing multiple cis-glycosidic linkages. Nat. Commun. 2016, 7, 12482. [Google Scholar] [CrossRef]
- Hurevich, M.; Seeberger, P.H. Automated glycopeptide assembly by combined solid-phase peptide and oligosaccharide synthesis. Chem. Commun. 2014, 50, 1851–1853. [Google Scholar] [CrossRef]
- Kandasamy, J.; Hurevich, M.; Seeberger, P.H. Automated solid phase synthesis of oligoarabinofuranosides. Chem. Commun. 2013, 49, 4453–4455. [Google Scholar] [CrossRef] [Green Version]
- Eller, S.; Collot, M.; Yin, J.; Hahm, H.S.; Seeberger, P.H. Automated solid-phase synthesis of chondroitin sulfate glycosaminoglycans. Angew. Chem. Int. Ed. 2013, 52, 5858–5861. [Google Scholar] [CrossRef] [PubMed]
- Kröck, L.; Esposito, D.; Castagner, B.; Wang, C.-C.; Bindschädler, P.; Seeberger, P.H. Streamlined access to conjugation-ready glycans by automated synthesis. Chem. Sci. 2012, 3, 1617–1622. [Google Scholar] [CrossRef]
- Tanaka, H.; Nishiura, Y.; Takahashi, T. Stereoselective synthesis of oligo-α-(2,8)-sialic acids. J. Am. Chem. Soc. 2006, 128, 7124–7125. [Google Scholar] [CrossRef] [PubMed]
- Crich, D.; Li, W. O-sialylation with N-acetyl-5-N,4-O-carbonyl-protected thiosialoside donors in dichloromethane: Facile and selective cleavage of the oxazolidinone ring. J. Org. Chem. 2007, 72, 2387–2391. [Google Scholar] [CrossRef] [PubMed]
- Crich, D.; Wu, B. Stereoselective iterative one-pot synthesis of N-glycolylneuraminic acid-containing oligosaccharides. Org. Lett. 2008, 10, 4033–4035. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Tanikawa, S.; Saito, R.; Sasaki, K. β-stereoselective mannosylation using 2,6-lactones. J. Am. Chem. Soc. 2016, 138, 14840–14843. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.-T.; Lin, Y.-C.; Lee, M.-N.; Chen, Y.-L.; Wang, M.-S.; Lai, C.-C. Biochemical characterization and substrate specificity of the gene cluster for biosyntheses of K-252a and its analogs by in vitro heterologous expression system of Escherichia coli. Mol. BioSyst. 2009, 5, 1192–1203. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, C.; Blanchard, S.; Thorson, J.S. Deciphering indolocarbazole and enediyne aminodideoxypentose biosynthesis through comparative genomics: Insights from the AT2433 biosynthetic locus. Chem. Biol. 2006, 13, 733–743. [Google Scholar] [CrossRef]
- Sánchez, C.; Méndez, C.; Salas, J.A. Indolocarbazole natural products: Occurrence, biosynthesis, and biological activity. Nat. Prod. Rep. 2006, 23, 1007–1045. [Google Scholar] [CrossRef]
- Zhang, C.; Albermann, C.; Fu, X.; Peters, N.R.; Chisholm, J.D.; Zhang, G.; Gilbert, E.J.; Wang, P.G.; Van Vranken, D.L.; Thorson, J.S. RebG- and RebM-catalyzed indolocarbazole diversification. ChemBioChem 2006, 7, 795–804. [Google Scholar] [CrossRef]
- Salas, A.P.; Zhu, L.; Sánchez, C.; Braña, A.F.; Rohr, J.; Méndez, C.; Salas, J.A. Deciphering the late steps in the biosynthesis of the anti-tumour indolocarbazole staurosporine: Sugar donor substrate flexibility of the StaG glycosyltransferase. Mol. Microbiol. 2005, 58, 17–27. [Google Scholar] [CrossRef]
- Hyun, C.-G.; Bililign, T.; Liao, J.; Thorson, J.S. The biosynthesis of indolocarbazoles in a heterologous E. coli host. ChemBioChem 2003, 4, 114–117. [Google Scholar] [CrossRef]
- Sánchez, C.; Butovich, I.A.; Braña, A.F.; Rohr, J.; Méndez, C.; Salas, J.A. The biosynthetic gene cluster for the antitumor rebeccamycin: Characterization and generation of indolocarbazole derivatives. Chem. Biol. 2002, 9, 519–531. [Google Scholar] [CrossRef]
- Ohuchi, T.; Ikeda-Araki, A.; Watanabe-Sakamoto, A.; Kojiri, K.; Nagashima, M.; Okanishi, M.; Suda, H. Cloning and expression of a gene encoding N-glycosyltransferase (ngt) from Saccharothrix aerocolonigenes ATCC39243. J. Antibiot. 2000, 53, 393–403. [Google Scholar] [CrossRef]
- Guo, Z.; Li, J.; Qin, H.; Wang, M.; Lv, X.; Li, X.; Chen, Y. Biosynthesis of the carbamoylated d-gulosamine moiety of streptothricins: Involvement of a guanidino-N-glycosyltransferase and an N-acetyl-d-gulosamine deacetylase. Angew. Chem. Int. Ed. 2015, 54, 5175–5178. [Google Scholar] [CrossRef]
- Gawthorne, J.A.; Tan, N.Y.; Bailey, U.-M.; Davis, M.R.; Wong, L.W.; Naidu, R.; Fox, K.L.; Jennings, M.P.; Schulz, B.L. Selection against glycosylation sites in potential target proteins of the general HMWC N-glycosyltransferase in Haemophilus influenzae. Biochem. Biophys. Res. Commun. 2014, 445, 633–638. [Google Scholar] [CrossRef]
- Naegeli, A.; Lin, C.-W.; Aebi, M.; Michaud, G.; Darbre, T.; Reymond, J.-L.; Schubert, M.; Lizak, C. Substrate specificity of cytoplasmic N-glycosyltransferase. J. Biol. Chem. 2014, 289, 24521–24532. [Google Scholar] [CrossRef]
- Choi, K.-J.; Grass, S.; Paek, S.; St. Geme, J.W., III; Yeo, H.-J. The Actinobacillus pleuropneumoniae HMW1C-like glycosyltransferase mediates N-linked glycosylation of the Haemophilus influenzae HMW1 adhesin. PLoS ONE 2011, 5, e15888. [Google Scholar] [CrossRef]
- Grass, S.; Lichti, C.F.; Townsend, R.R.; Gross, J.; St. Geme, J.W., III. The Haemophilus influenzae HMW1C protein is a glycosyltransferase that transfers hexose residues to asparagine sites in the HMW1 adhesin. PLoS Pathog. 2010, 6, e1000919. [Google Scholar] [CrossRef]
- Zhao, P.; Bai, L.; Ma, J.; Zeng, Y.; Li, L.; Zhang, Y.; Lu, C.; Dai, H.; Wu, Z.; Li, Y.; et al. Amide N-glycosylation by Asm25, an N-glycosyltransferase of ansamitocins. Chem. Biol. 2008, 15, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Magarvey, N.A.; Haltli, B.; He, M.; Greenstein, M.; Hucul, J.A. Biosynthetic pathway for mannopeptimycins, lipoglycopeptide antibiotics active against drug-resistant gram-positive pathogens. Antimicrob. Agents Chemother. 2006, 50, 2167–2177. [Google Scholar] [CrossRef]
- Chen, D.; Chen, R.; Wang, R.; Li, J.; Xie, K.; Bian, C.; Sun, L.; Zhang, X.; Liu, J.; Yang, L.; et al. Probing the catalytic promiscuity of a regio- and stereospecific C-glycosyltransferase from Mangifera indica. Angew. Chem. Int. Ed. 2015, 54, 12678–12682. [Google Scholar] [CrossRef]
- Foshag, D.; Campbell, C.; Pawelek, P.D. The C-glycosyltransferase IroB from pathogenic Escherichia coli: Identification of residues required for efficient catalysis. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2014, 1844, 1619–1630. [Google Scholar] [CrossRef]
- Gutmann, A.; Krump, C.; Bungaruang, L.; Nidetzky, B. A two-step O- to C-glycosidic bond rearrangement using complementary glycosyltransferase activities. Chem. Commun. 2014, 50, 5465–5468. [Google Scholar] [CrossRef]
- Panek, A.; Pietrow, O.; Synowiecki, J.; Filipkowski, P. Immobilization on magnetic nanoparticles of the recombinant trehalose synthase from Deinococcus geothermalis. Food Bioprod. Process. 2013, 91, 632–637. [Google Scholar] [CrossRef]
- Tsai, T.-I.; Lee, H.-Y.; Chang, S.-H.; Wang, C.-H.; Tu, Y.-C.; Lin, Y.-C.; Hwang, D.-R.; Wu, C.-Y.; Wong, C.-H. Effective sugar nucleotide regeneration for the large-scale enzymatic synthesis of Globo H and SSEA4. J. Am. Chem. Soc. 2013, 135, 14831–14839. [Google Scholar] [CrossRef]
- Gutmann, A.; Nidetzky, B. Switching between O- and C-glycosyltransferase through exchange of active-site motifs. Angew. Chem. Int. Ed. 2012, 51, 12879–12883. [Google Scholar] [CrossRef]
- Härle, J.; Günther, S.; Lauinger, B.; Weber, M.; Kammerer, B.; Zechel, D.L.; Luzhetskyy, A.; Bechthold, A. Rational design of an aryl-C-glycoside catalyst from a natural product O-glycosyltransferase. Chem. Biol. 2011, 18, 520–530. [Google Scholar]
- Mittler, M.; Bechthold, A.; Schulz, G.E. Structure and action of the C-C bond-forming glycosyltransferase UrdGT2 involved in the biosynthesis of the antibiotic urdamycin. J. Mol. Biol. 2007, 372, 67–76. [Google Scholar] [CrossRef]
- Baig, I.; Kharel, M.; Kobylyanskyy, A.; Zhu, L.; Rebets, Y.; Ostash, B.; Luzhetskyy, A.; Bechthold, A.; Fedorenko, V.A.; Rohr, J. On the acceptor substrate of C-glycosyltransferase UrdGT2: Three prejadomycin C-glycosides from an engineered mutant of Streptomyces globisporus 1912 ΔIndE(UrdGT2). Angew. Chem. Int. Ed. 2006, 45, 7842–7846. [Google Scholar] [CrossRef]
- Liu, T.; Kharel, M.K.; Fischer, C.; McCormick, A.; Rohr, J. Inactivation of gilGT, encoding a C-glycosyltransferase, and gilOIII, encoding a P450 enzyme, allows the details of the late biosynthetic pathway to gilvocarcin V to be delineated. ChemBioChem 2006, 7, 1070–1077. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Lin, H.; Liu, D.R.; Walsh, C.T. In vitro characterization of IroB, a pathogen-associated C-glycosyltransferase. Proc. Natl. Acad. Sci. USA 2005, 102, 571–576. [Google Scholar] [CrossRef]
- Bililign, T.; Hyun, C.-G.; Williams, J.S.; Czisny, A.M.; Thorson, J.S. The hedamycin locus implicates a novel aromatic PKS priming mechanism. Chem. Biol. 2004, 11, 959–969. [Google Scholar] [CrossRef]
- Kopycki, J.; Wieduwild, E.; Kohlschmidt, J.; Brandt, W.; Stepanova, A.N.; Alonso, J.M.; Pedras, M.; Soledade, C.; Abel, S.; Grubb, C.D.; et al. Kinetic analysis of Arabidopsis glucosyltransferase UGT74B1 illustrates a general mechanism by which enzymes can escape product inhibition. Biochem. J. 2013, 450, 37–46. [Google Scholar] [CrossRef]
- Wang, H.; van der Donk, W.A. Substrate selectivity of the sublancin S-glycosyltransferase. J. Am. Chem. Soc. 2011, 133, 16394–16397. [Google Scholar] [CrossRef]
- Almendros, M.; Danalev, D.; François-Heude, M.; Loyer, P.; Legentil, L.; Nugier-Chauvin, C.; Daniellou, R.; Ferrières, V. Exploring the synthetic potency of the first furanothioglycoligase through original remote activation. Org. Biomol. Chem. 2011, 9, 8371–8378. [Google Scholar] [CrossRef] [Green Version]
- Douglas Grubb, C.; Zipp, B.J.; Ludwig-Müller, J.; Masuno, M.N.; Molinski, T.F.; Abel, S. Arabidopsis glucosyltransferase UGT74B1 functions in glucosinolate biosynthesis and auxin homeostasis. Plant J. 2004, 40, 893–908. [Google Scholar] [CrossRef]
- Calderon, A.D.; Zhou, J.; Guan, W.; Wu, Z.; Guo, Y.; Bai, J.; Li, Q.; Wang, P.G.; Fang, J.; Li, L. An enzymatic strategy to asymmetrically branched N-glycans. Org. Biomol. Chem. 2017, 15, 7258–7262. [Google Scholar] [CrossRef]
- Gagarinov, I.A.; Li, T.; Toraño, J.S.; Caval, T.; Srivastava, A.D.; Kruijtzer, J.A.W.; Heck, A.J.R.; Boons, G.-J. Chemoenzymatic approach for the preparation of asymmetric bi-, tri-, and tetra-antennary N-glycans from a common precursor. J. Am. Chem. Soc. 2017, 139, 1011–1018. [Google Scholar] [CrossRef]
- Xiao, Z.; Guo, Y.; Liu, Y.; Li, L.; Zhang, Q.; Wen, L.; Wang, X.; Kondengaden, S.M.; Wu, Z.; Zhou, J.; et al. Chemoenzymatic synthesis of a library of human milk oligosaccharides. J. Org. Chem. 2016, 81, 5851–5865. [Google Scholar] [CrossRef]
- Zhang, C.; Griffith, B.R.; Fu, Q.; Albermann, C.; Fu, X.; Lee, I.-K.; Li, L.; Thorson, J.S. Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science 2006, 313, 1291–1294. [Google Scholar] [CrossRef]
- Gantt, R.W.; Peltier-Pain, P.; Cournoyer, W.J.; Thorson, J.S. Using simple donors to drive the equilibria of glycosyltransferase-catalyzed reactions. Nat. Chem. Biol. 2011, 7, 685. [Google Scholar] [CrossRef]
- Gantt, R.W.; Thorson, J.S. Chapter seventeen—High-throughput colorimetric assays for nucleotide sugar formation and glycosyl transfer. In Methods in Enzymology; Hopwood, D.A., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 516, pp. 345–360. [Google Scholar]
- Peltier-Pain, P.; Marchillo, K.; Zhou, M.; Andes, D.R.; Thorson, J.S. Natural product disaccharide engineering through tandem glycosyltransferase catalysis reversibility and neoglycosylation. Org. Lett. 2012, 14, 5086–5089. [Google Scholar] [CrossRef]
- Zhou, M.; Hamza, A.; Zhan, C.-G.; Thorson, J.S. Assessing the regioselectivity of OleD-catalyzed glycosylation with a diverse set of acceptors. J. Nat. Prod. 2013, 76, 279–286. [Google Scholar] [CrossRef]
- Gantt, R.W.; Peltier-Pain, P.; Singh, S.; Zhou, M.; Thorson, J.S. Broadening the scope of glycosyltransferase-catalyzed sugar nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2013, 110, 7648–7653. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, P.S.; Mansell, R.L. Isolation and partial characterization of three glucosyl transferases involved in the biosynthesis of flavonol triglucosides in Pisum sativum L. Archives of Biochemistry and Biophysics 1982, 213, 434–443. [Google Scholar] [CrossRef]
- Shearer, J.; Graham, T.E. Novel aspects of skeletal muscle glycogen and its regulation during rest and exercise. Exerc. Sport Sci. Rev. 2004, 32, 120–126. [Google Scholar] [CrossRef]
- Mavrovouniotis, M.L. Estimation of standard Gibbs energy changes of biotransformations. J. Biol. Chem. 1991, 266, 14440–14445. [Google Scholar]
- Jankowski, M.D.; Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 2008, 95, 1487–1499. [Google Scholar] [CrossRef]
- Bar-Even, A.; Flamholz, A.; Davidi, D.; Noor, E.; Milo, R.; Lubling, Y. An integrated open framework for thermodynamics of reactions that combines accuracy and coverage. Bioinformatics 2012, 28, 2037–2044. [Google Scholar] [Green Version]
- Noor, E.; Haraldsdóttir, H.S.; Milo, R.; Fleming, R.M.T. Consistent estimation of Gibbs energy using component contributions. PLoS Comput. Biol. 2013, 9, e1003098. [Google Scholar] [CrossRef]
- Goldberg, R.N.; Bhat, T.N.; Tewari, Y.B. Thermodynamics of enzyme-catalyzed reactions—A database for quantitative biochemistry. Bioinformatics 2004, 20, 2874–2877. [Google Scholar] [CrossRef]
- Minakami, S.; Yoshikawa, H. Thermodynamic considerations on erythrocyte glycolysis. Biochem. Biophys. Res. Commun. 1965, 18, 345–349. [Google Scholar] [CrossRef]
- Held, C.; Sadowski, G. Thermodynamics of bioreactions. Annual Rev. Chem. Biomol. Eng. 2016, 7, 395–414. [Google Scholar] [CrossRef]
- Ozaki, S.-I.; Imai, H.; Iwakiri, T.; Sato, T.; Shimoda, K.; Nakayama, T.; Hamada, H. Regioselective glucosidation of trans-resveratrol in Escherichia coli expressing glucosyltransferase from Phytolacca americana. Biotechnol. Lett. 2012, 34, 475–481. [Google Scholar] [CrossRef]
- Yahyaa, M.; Davidovich-Rikanati, R.; Eyal, Y.; Sheachter, A.; Marzouk, S.; Lewinsohn, E.; Ibdah, M. Identification and characterization of UDP-glucose: Phloretin 4′-O-glycosyltransferase from Malus x domestica Borkh. Phytochemistry 2016, 130, 47–55. [Google Scholar] [CrossRef]
- Zhang, T.; Liang, J.; Wang, P.; Xu, Y.; Wang, Y.; Wei, X.; Fan, M. Purification and characterization of a novel phloretin-2′-O-glycosyltransferase favoring phloridzin biosynthesis. Sci. Rep. 2016, 6, 35274. [Google Scholar] [CrossRef]
- Lim, E.-K.; Doucet, C.J.; Li, Y.; Elias, L.; Worrall, D.; Spencer, S.P.; Ross, J.; Bowles, D.J. The activity of Arabidopsis glycosyltransferases toward salicylic acid, 4-hydroxybenzoic acid, and other benzoates. J. Biol. Chem. 2002, 277, 586–592. [Google Scholar] [CrossRef]
- Tadera, K.; Yagi, F.; Kobayashi, A. Specificity of a particulate glycosyltransferase in seedlings of Pisum sativum L. Which catalyzes the formation of 5′-O-(β-d-glucopyranosyl)pyridoxine. J. Nutr. Sci. Vitaminol. 1982, 28, 359–366. [Google Scholar] [CrossRef]
- Lin, J.-S.; Huang, X.-X.; Li, Q.; Cao, Y.; Bao, Y.; Meng, X.-F.; Li, Y.-J.; Fu, C.; Hou, B.-K. UDP-glycosyltransferase 72B1 catalyzes the glucose conjugation of monolignols and is essential for the normal cell wall lignification in Arabidopsis thaliana. Plant J. 2016, 88, 26–42. [Google Scholar] [CrossRef]
- Ibrahim, R.K.; Grisebach, H. Purification and properties of UDP-glucose: Coniferyl alcohol glucosyltransferase from suspension cultures of Paul’s scarlet rose. Arch. Biochem. Biophys. 1976, 176, 700–708. [Google Scholar] [CrossRef]
- Hyung Ko, J.; Gyu Kim, B.; Joong-Hoon, A. Glycosylation of flavonoids with a glycosyltransferase from Bacillus cereus. FEMS Microbiol. Lett. 2006, 258, 263–268. [Google Scholar] [CrossRef]
- Ostrowski, M.; Hetmann, A.; Jakubowska, A. Indole-3-acetic acid UDP-glucosyltransferase from immature seeds of pea is involved in modification of glycoproteins. Phytochemistry 2015, 117, 25–33. [Google Scholar] [CrossRef]
- Tahara, K.; Nishiguchi, M.; Frolov, A.; Mittasch, J.; Milkowski, C. Identification of UDP glucosyltransferases from the aluminum-resistant tree Eucalyptus camaldulensis forming β-glucogallin, the precursor of hydrolyzable tannins. Phytochemistry 2018, 152, 154–161. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, Y.; Xu, J.; Wang, X.; Shen, C.; Zhang, Y.; Liu, X.; Yu, B.; Zhang, J. Molecular cloning and expression of a glycosyltransferase from Bacillus subtilis for modification of morin and related polyphenols. Biotechnol. Lett. 2017, 39, 1229–1235. [Google Scholar] [CrossRef]
- Marcinek, H.; Weyler, W.; Deus-Neumann, B.; Zenk, M.H. Indoxyl-UDPG-glucosyltransferase from Baphicacanthus cusia. Phytochemistry 2000, 53, 201–207. [Google Scholar] [CrossRef]
- Mandal, S.S.; Liao, G.; Guo, Z. Chemical synthesis of the tumor-associated Globo H antigen. RSC Adv. 2015, 5, 23311–23319. [Google Scholar] [CrossRef]
- Frear, D.S. Herbicide metabolism in plants—I: Purification and properties of UDP-glucose: arylamine N-glucosyl-transferase from soybean. Phytochemistry 1968, 7, 381–390. [Google Scholar] [CrossRef]
- Martin, R.C.; Mok, M.C.; Mok, D.W.S. Isolation of a cytokinin gene, ZOG1, encoding zeatin O-glucosyltransferase from Phaseolus lunatus. Proc. Natl. Acad. Sci. USA 1999, 96, 284–289. [Google Scholar] [CrossRef]
- Landmann, C.; Fink, B.; Schwab, W. FaGT2: A multifunctional enzyme from strawberry (Fragaria x ananassa) fruits involved in the metabolism of natural and xenobiotic compounds. Planta 2007, 226, 417–428. [Google Scholar] [CrossRef]
- Lunkenbein, S.; Bellido, M.; Aharoni, A.; Salentijn, E.M.J.; Kaldenhoff, R.; Coiner, H.A.; Muñoz-Blanco, J.; Schwab, W. Cinnamate metabolism in ripening fruit. Characterization of a UDP-glucose: cinnamate glucosyltransferase from strawberry. Plant Physiol. 2006, 140, 1047–1058. [Google Scholar] [CrossRef]
- Trobo-Maseda, L.; Orrego, A.H.; Moreno-Pérez, S.; Fernández-Lorente, G.; Guisan, J.M.; Rocha-Martin, J. Stabilization of multimeric sucrose synthase from Acidithiobacillus caldus via immobilization and post-immobilization techniques for synthesis of UDP-glucose. Appl. Microbiol. Biotechnol. 2018, 102, 773–787. [Google Scholar] [CrossRef]
- Dewitte, G.; Walmagh, M.; Diricks, M.; Lepak, A.; Gutmann, A.; Nidetzky, B.; Desmet, T. Screening of recombinant glycosyltransferases reveals the broad acceptor specificity of stevia UGT-76G1. J. Biotechnol. 2016, 233, 49–55. [Google Scholar] [CrossRef]
- Gutmann, A.; Nidetzky, B. Unlocking the potential of leloir glycosyltransferases for applied biocatalysis: Efficient synthesis of uridine 5′-diphosphate-glucose by sucrose synthase. Adv. Synth. Catal. 2016, 358, 3600–3609. [Google Scholar] [CrossRef]
- Ryu, S.-I.; Park, C.-S.; Cha, J.; Woo, E.-J.; Lee, S.-B. A novel trehalose-synthesizing glycosyltransferase from Pyrococcus horikoshii: Molecular cloning and characterization. Biochem. Biophys. Res. Commun. 2005, 329, 429–436. [Google Scholar] [CrossRef]
- Tewari, Y.B.; Goldberg, R.N. Thermodynamics of hydrolysis of disaccharides: Lactulose, α-d-melibiose, palatinose, d-trehalose, d-turanose and 3-O-β-d-galactopyranosyl-d-arabinose. Biophys. Chem. 1991, 40, 59–67. [Google Scholar] [CrossRef]
- Qu, Q.; Lee, S.-J.; Boos, W. TreT, a novel trehalose glycosyltransferring synthase of the hyperthermophilic archaeon Thermococcus litoralis. J. Biol. Chemistry 2004, 279, 47890–47897. [Google Scholar] [CrossRef]
- Kouril, T.; Zaparty, M.; Marrero, J.; Brinkmann, H.; Siebers, B. A novel trehalose synthesizing pathway in the hyperthermophilic crenarchaeon Thermoproteus tenax: The unidirectional tret pathway. Arch. Microbiol. 2008, 190, 355. [Google Scholar] [CrossRef]
- Tian, C.; Yang, J.; Zeng, Y.; Zhang, T.; Zhou, Y.; Men, Y.; You, C.; Zhu, Y.; Sun, Y. Biosynthesis of raffinose and stachyose from sucrose via an in vitro multienzyme system. Appl. Environ. Microbiol. 2019, 85, e02306–e02318. [Google Scholar] [CrossRef]
- Resnick, S.M.; Zehnder, A.J.B. In vitro atp regeneration from polyphosphate and AMP by polyphosphate: AMP phosphotransferase and adenylate kinase from Acinetobacter johnsonii 210A. Appl. Environ. Microbiol. 2000, 66, 2045–2051. [Google Scholar] [CrossRef]
- Crans, D.C.; Whitesides, G.M. A convenient synthesis of disodium acetyl phosphate for use in in situ ATP cofactor regeneration. J. Org. Chem. 1983, 48, 3130–3132. [Google Scholar] [CrossRef]
- Tasnádi, G.; Jud, W.; Hall, M.; Baldenius, K.; Ditrich, K.; Faber, K. Evaluation of natural and synthetic phosphate donors for the improved enzymatic synthesis of phosphate monoesters. Adv. Synth. Catal. 2018, 360, 2394–2401. [Google Scholar] [CrossRef]
- Kulmer, S.T.; Gutmann, A.; Lemmerer, M.; Nidetzky, B. Biocatalytic cascade of polyphosphate kinase and sucrose synthase for synthesis of nucleotide-activated derivatives of glucose. Adv. Synth. Catal. 2017, 359, 292–301. [Google Scholar] [CrossRef]
- Andexer, J.N.; Richter, M. Emerging enzymes for atp regeneration in biocatalytic processes. ChemBioChem 2015, 16, 380–386. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, J.; Chen, X.; Wang, P.G. Combined biosynthetic pathway for de novo production of udp-galactose: Catalysis with multiple enzymes immobilized on agarose beads. ChemBioChem 2002, 3, 348–355. [Google Scholar] [CrossRef]
- Murata, K.; Uchida, T.; Kato, J.; Chibata, I. Polyphosphate kinase: Distribution, some properties and its application as an atp regeneration system. Agric. Biol. Chem. 1988, 52, 1471–1477. [Google Scholar]
- Restiawaty, E.; Iwasa, Y.; Maya, S.; Honda, K.; Omasa, T.; Hirota, R.; Kuroda, A.; Ohtake, H. Feasibility of thermophilic adenosine triphosphate-regeneration system using Thermus thermophilus polyphosphate kinase. Process. Biochem. 2011, 46, 1747–1752. [Google Scholar] [CrossRef]
- Sato, M.; Masuda, Y.; Kirimura, K.; Kino, K. Thermostable ATP regeneration system using polyphosphate kinase from thermosynechococcus elongatus BP-1 for d-amino acid dipeptide synthesis. J. Biosci. Bioeng. 2007, 103, 179–184. [Google Scholar] [CrossRef]
- Iwamoto, S.; Motomura, K.; Shinoda, Y.; Urata, M.; Kato, J.; Takiguchi, N.; Ohtake, H.; Hirota, R.; Kuroda, A. Use of an Escherichia coli recombinant producing thermostable polyphosphate kinase as an ATP regenerator to produce fructose 1,6-diphosphate. Appl. Environ. Microbiol. 2007, 73, 5676–5678. [Google Scholar] [CrossRef]
- 1Shiba, T.; Tsutsumi, K.; Ishige, K.; Noguchi, T. Inorganic polyphosphate and polyphosphate kinase: Their novel biological functions and applications. Biochemistry (Moscow) 2000, 65, 315–323. [Google Scholar]
- Noguchi, T.; Shiba, T. Use of Escherichia coli polyphosphate kinase for oligosaccharide synthesis. Biosci. Biotechnol. Biochem. 1998, 62, 1594–1596. [Google Scholar] [CrossRef]
- Nahálka, J.; Pätoprstý, V. Enzymatic synthesis of sialylation substrates powered by a novel polyphosphate kinase (PPK3). Org. Biomol. Chem. 2009, 7, 1778–1780. [Google Scholar] [CrossRef]
- Lee, S.-G.; Lee, J.-O.; Yi, J.-K.; Kim, B.-G. Production of cytidine 5′-monophosphate N-acetylneuraminic acid using recombinant Escherichia coli as a biocatalyst. Biotechnol. Bioeng. 2002, 80, 516–524. [Google Scholar] [CrossRef]
- Rupprath, C.; Kopp, M.; Hirtz, D.; Müller, R.; Elling, L. An enzyme module system for in situ regeneration of deoxythymidine 5′-diphosphate (dTDP)-activated deoxy sugars. Adv. Synt. Catal. 2007, 349, 1489–1496. [Google Scholar] [CrossRef]
- Fischöder, T.; Wahl, C.; Zerhusen, C.; Elling, L. Repetitive batch mode facilitates enzymatic synthesis of the nucleotide sugars UDP-Gal, UDP-GlcNAc, and UDP-GalNAc on a multi-gram scale. Biotechnol. J. 2019, 14. [Google Scholar] [CrossRef]
- Wen, L.; Zheng, Y.; Li, T.; Wang, P.G. Enzymatic synthesis of 3-deoxy-d-manno-octulosonic acid (KDO) and its application for LPS assembly. Bioorg. Med. Chem. Lett. 2016, 26, 2825–2828. [Google Scholar] [CrossRef]
- Lee, J.-H.; Chung, S.-W.; Lee, H.-J.; Jang, K.-S.; Lee, S.-G.; Kim, B.-G. Optimization of the enzymatic one pot reaction for the synthesis of uridine 5′-diphosphogalactose. Bioprocess. Biosyst. Eng. 2009, 33, 71. [Google Scholar] [CrossRef]
- Unverzagt, C.; Kunz, H.; Paulson, J.C. High-efficiency synthesis of sialyloligosaccharides and sialoglycopeptides. J. Am. Chem. Soc. 1990, 112, 9308–9309. [Google Scholar] [CrossRef]
- Hirschbein, B.L.; Mazenod, F.P.; Whitesides, G.M. Synthesis of phosphoenolypyruvate and its use in ATP cofactor regeneration. J. Org. Chem. 1982, 47, 3765–3766. [Google Scholar] [CrossRef]
- Yu, H.; Santra, A.; Li, Y.; McArthur, J.B.; Ghosh, T.; Yang, X.; Wang, P.G.; Chen, X. Streamlined chemoenzymatic total synthesis of prioritized ganglioside cancer antigens. Org. Biomol. Chem. 2018, 16, 4076–4080. [Google Scholar] [CrossRef] [PubMed]
- Muthana, M.M.; Qu, J.; Xue, M.; Klyuchnik, T.; Siu, A.; Li, Y.; Zhang, L.; Yu, H.; Li, L.; Wang, P.G.; et al. Improved one-pot multienzyme (OPME) systems for synthesizing UDP-uronic acids and glucuronides. Chem. Commun. 2015, 51, 4595–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, J.A.; Ahmed, R.A.; Tanner, M.E. Efficient chemoenzymatic synthesis of ADP-d-glycero-β-d-manno-heptose and a mechanistic study of ADP-l-glycero-d-manno-heptose 6-epimerase. Org. Lett. 2005, 7, 2457–2460. [Google Scholar] [CrossRef] [PubMed]
- Alissandratos, A.; Caron, K.; Loan, T.D.; Hennessy, J.E.; Easton, C.J. ATP recycling with cell lysate for enzyme-catalyzed chemical synthesis, protein expression and PCR. ACS Chem. Biol. 2016, 11, 3289–3293. [Google Scholar] [CrossRef]
- Wong, C.H.; Haynie, S.L.; Whitesides, G.M. Enzyme-catalyzed synthesis of N-acetyllactosamine with in situ regeneration of uridine 5′-diphosphate glucose and uridine 5′-diphosphate galactose. J. Org. Chem. 1982, 47, 5416–5418. [Google Scholar] [CrossRef]
- Tomoike, F.; Nakagawa, N.; Kuramitsu, S.; Masui, R. A single amino acid limits the substrate specificity of Thermus thermophilus uridine-cytidine kinase to cytidine. Biochemistry 2011, 50, 4597–4607. [Google Scholar] [CrossRef]
- Loan, T.D.; Easton, C.J.; Alissandratos, A. Recombinant cell-lysate-catalysed synthesis of uridine-5′-triphosphate from nucleobase and ribose, and without addition of ATP. New Biotechnol. 2019, 49, 104–111. [Google Scholar] [CrossRef]
- Scism, R.A.; Bachmann, B.O. Five-component cascade synthesis of nucleotide analogues in an engineered self-immobilized enzyme aggregate. ChemBioChem 2010, 11, 67–70. [Google Scholar] [CrossRef]
- Fernández-Lucas, J. Multienzymatic synthesis of nucleic acid derivatives: A general perspective. Appl. Microbiol. Biotechnol. 2015, 99, 4615–4627. [Google Scholar] [CrossRef]
- Glaser, L.; Brown, D.H. The synthesis of chitin in cell-free extracts of Neurospora crassa. J. Biol. Chem. 1957, 228, 729–742. [Google Scholar]
- Lougheed, B.; Ly, H.D.; Wakarchuk, W.W.; Withers, S.G. Glycosyl fluorides can function as substrates for nucleotide phosphosugar-dependent glycosyltransferases. J. Biol. Chem. 1999, 274, 37717–37722. [Google Scholar] [CrossRef] [PubMed]
- Lepak, A.; Gutmann, A.; Nidetzky, B. β-glucosyl fluoride as reverse reaction donor substrate and mechanistic probe of inverting sugar nucleotide-dependent glycosyltransferases. ACS Catal. 2018, 8, 9148–9153. [Google Scholar] [CrossRef]
- Lairson, L.L.; Wakarchuk, W.W.; Withers, S.G. Alternative donor substrates for inverting and retaining glycosyltransferases. Chem. Commun. 2007, 4, 365–367. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Z.; Duan, Q.; Li, X. Glycopeptide synthesis on an ionic liquid support. Org. Lett. 2014, 16, 3008–3011. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.R.; Shaaban, K.A.; Zhang, J.; Cao, H.; Phillips, G.N.; Thorson, J.S. OleD Loki as a catalyst for tertiary amine and hydroxamate glycosylation. ChemBioChem 2017, 18, 363–367. [Google Scholar] [CrossRef]
- Lee, A.A.; Chen, Y.C.S.; Ekalestari, E.; Ho, S.Y.; Hsu, N.S.; Kuo, T.F.; Wang, T.S.A. Facile and versatile chemoenzymatic synthesis of enterobactin analogues and applications in bacterial detection. Angew. Chem. Int. Ed. 2016, 55, 12338–12342. [Google Scholar] [CrossRef]
- Burkhart, F.; Zhang, Z.; Wacowich-Sgarbi, S.; Wong, C.H. Synthesis of the Globo H hexasaccharide using the programmable reactivity-based one-pot strategy. Angew. Chem. Int. Ed. 2001, 40, 1274–1277. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Thayer, D.A.; Chang, A.Y.; Best, M.D.; Hoffmann, J.; Head, S.; Wong, C.-H. Carbohydrate microarray for profiling the antibodies interacting with Globo H tumor antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 15–20. [Google Scholar] [CrossRef]
- Su, D.M.; Eguchi, H.; Yi, W.; Li, L.; Wang, P.G.; Xia, C. Enzymatic synthesis of tumor-associated carbohydrate antigen Globo-H hexasaccharide. Org. Lett. 2008, 10, 1009–1012. [Google Scholar] [CrossRef]
- Yu, H.; Lau, K.; Li, Y.; Sugiarto, G.; Chen, X. One-pot multienzyme synthesis of Lewis x and sialyl Lewis x antigens. Curr. Protoc. Chem. Biol. 2012, 4, 233–247. [Google Scholar]
- Nishimura, S.-I.; Yamada, K. Transfer of ganglioside GM3 oligosaccharide from a water soluble polymer to ceramide by ceramide glycanase. A novel approach for the chemical-enzymatic synthesis of glycosphingolipids. J. Am. Chem. Soc. 1997, 119, 10555–10556. [Google Scholar] [CrossRef]
- Huang, X.; Witte, K.L.; Bergbreiter, D.E.; Wong, C.-H. Homogenous enzymatic synthesis using a thermo-responsive water-soluble polymer support. Adv. Synt. Catal. 2001, 343, 675–681. [Google Scholar] [CrossRef]
- Jaipuri, F.A.; Pohl, N.L. Toward solution-phase automated iterative synthesis: Fluorous-tag assisted solution-phase synthesis of linear and branched mannose oligomers. Org. Biomol. Chem. 2008, 6, 2686–2691. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Jing, Y.; Huang, X. Fluorous-assisted one-pot oligosaccharide synthesis. Eur. J. Org. Chem. 2010, 2010, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Pohl, N.L.B. A fluorous phosphate protecting group with applications to carbohydrate synthesis. Org. Lett. 2011, 13, 1824–1827. [Google Scholar] [CrossRef]
- Carrel, F.R.; Geyer, K.; Codée, J.D.C.; Seeberger, P.H. Oligosaccharide synthesis in microreactors. Org. Lett. 2007, 9, 2285–2288. [Google Scholar] [CrossRef]
- Carrel, F.R.; Seeberger, P.H. Cap-and-tag solid phase oligosaccharide synthesis. J. Org. Chem. 2008, 73, 2058–2065. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, W.; Zhang, Y.; Curran, D.P.; Liu, G. Synthesis and applications of a light-fluorous glycosyl donor. J. Org. Chem. 2009, 74, 2594–2597. [Google Scholar] [CrossRef]
- Huang, W.; Gao, Q.; Boons, G.-J. Assembly of a complex branched oligosaccharide by combining fluorous-supported synthesis and stereoselective glycosylations using anomeric sulfonium ions. Chem. Eur. J. 2015, 21, 12920–12926. [Google Scholar] [CrossRef]
- Macchione, G.; de Paz, J.L.; Nieto, P.M. Synthesis of hyaluronic acid oligosaccharides and exploration of a fluorous-assisted approach. Carbohydr. Res. 2014, 394, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Chai, Y.-H.; Feng, Y.-L.; Wu, J.-J.; Deng, C.-Q.; Liu, A.-Y.; Zhang, Q. Recyclable benzyl-type fluorous tags: Preparation and application in oligosaccharide synthesis. Chin. Chem. Lett. 2017, 28, 1693–1700. [Google Scholar] [CrossRef]
- Hwang, J.; Yu, H.; Malekan, H.; Sugiarto, G.; Li, Y.; Qu, J.; Nguyen, V.; Wu, D.; Chen, X. Highly efficient one-pot multienzyme (OPME) synthesis of glycans with fluorous-tag assisted purification. Chem. Commun. 2014, 50, 3159–3162. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Tanimoto, Y.; Kawai, T.; Takahashi, T. A fluorous-assisted synthesis of oligosaccharides using a phenyl ether linker as a safety-catch linker. Tetrahedron 2011, 67, 10011–10016. [Google Scholar] [CrossRef]
- Zhu, H.; Wu, Z.; Gadi, M.R.; Wang, S.; Guo, Y.; Edmunds, G.; Guan, W.; Fang, J. Cation exchange assisted binding-elution strategy for enzymatic synthesis of human milk oligosaccharides (HMOS). Bioorg. Med. Chem. Lett. 2017, 27, 4285–4287. [Google Scholar] [CrossRef]
- Santra, A.; Li, Y.; Yu, H.; Slack, T.J.; Wang, P.G.; Chen, X. Highly efficient chemoenzymatic synthesis and facile purification of α-Gal pentasaccharyl ceramide Galα3nLc4βCer. Chem. Commun. 2017, 53, 8280–8283. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, C.; Gadi, M.R.; Gibbons, C.; Guo, Y.; Cao, X.; Edmunds, G.; Wang, S.; Liu, D.; Yu, J.; et al. Machine-driven enzymatic oligosaccharide synthesis by using a peptide synthesizer. Angew. Chem. Int. Ed. 2018, 57, 16638–16642. [Google Scholar] [CrossRef]
- Schuster, M.; Wang, P.; Paulson, J.C.; Wong, C.-H. Solid-phase chemical-enzymic synthesis of glycopeptides and oligosaccharides. J. Am. Chem. Soc. 1994, 116, 1135–1136. [Google Scholar] [CrossRef]
- Halcomb, R.L.; Huang, H.; Wong, C.-H. Solution- and solid-phase synthesis of inhibitors of H. pylori attachment and E-selectin-mediated leukocyte adhesion. J. Am. Chem. Soc. 1994, 116, 11315–11322. [Google Scholar] [CrossRef]
- Blixt, O.; Norberg, T. Enzymatic glycosylation of reducing oligosaccharides linked to a solid phase or a lipid via a cleavable squarate linker. Carbohydr. Res. 1999, 319, 80–91. [Google Scholar] [CrossRef]
- Houseman, B.T.; Mrksich, M. The role of ligand density in the enzymatic glycosylation of carbohydrates presented on self-assembled monolayers of alkanethiolates on gold. Angew. Chem. Int. Ed. 1999, 38, 782–785. [Google Scholar] [CrossRef]
- Yamada, K.; Fujita, E.; Nishimura, S.-I. High performance polymer supports for enzyme-assisted synthesis of glycoconjugates. Carbohydr. Res. 1997, 305, 443–461. [Google Scholar] [CrossRef]
- Yan, F.; Wakarchuk, W.W.; Gilbert, M.; Richards, J.C.; Whitfield, D.M. Polymer-supported and chemoenzymatic synthesis of the Neisseria meningitidis pentasaccharide: A methodological comparison. Carbohydr. Res. 2000, 328, 3–16. [Google Scholar] [CrossRef]
- Sears, P.; Wong, C.-H. Toward automated synthesis of oligosaccharides and glycoproteins. Science 2001, 291, 2344–2350. [Google Scholar] [CrossRef] [PubMed]
- Ivannikova, T.; Bintein, F.; Malleron, A.; Juliant, S.; Cerutti, M.; Harduin-Lepers, A.; Delannoy, P.; Augé, C.; Lubineau, A. Recombinant (2→3)-α-sialyltransferase immobilized on nickel-agarose for preparative synthesis of sialyl Lewisx and Lewisa precursor oligosaccharides. Carbohydr. Res. 2003, 338, 1153–1161. [Google Scholar] [CrossRef]
- Augé, C.; Fernandez-Fernandez, R.; Gautheron, C. The use of immobilised glycosyltransferases in the synthesis of sialyloligosaccharides. Carbohydr. Res. 1990, 200, 257–268. [Google Scholar] [CrossRef]
- Augé, C.; Gautheron, C. An efficient synthesis of cytidine monophospho-sialic acids with four immobilized enzymes. Tetrahedron Lett. 1988, 29, 789–790. [Google Scholar] [CrossRef]
- David, S.; Auge, C. Immobilized enzymes in preparative carbohydrate chemistry. Pure Appl. Chem. 1987, 59, 1501–1508. [Google Scholar] [CrossRef]
- Augé, C.; David, S.; Mathieu, C.; Gautheron, C. Synthesis with immobilized enzymes of two trisaccharides, one of them active as the determinant of a stage antigen. Tetrahedron Lett. 1984, 25, 1467–1470. [Google Scholar] [CrossRef]
- Chen, X.; Fang, J.; Zhang, J.; Liu, Z.; Shao, J.; Kowal, P.; Andreana, P.; Wang, P.G. Sugar nucleotide regeneration beads (superbeads): A versatile tool for the practical synthesis of oligosaccharides. J. Am. Chem. Soc. 2001, 123, 2081–2082. [Google Scholar] [CrossRef]
- Yu, C.-C.; Kuo, Y.-Y.; Liang, C.-F.; Chien, W.-T.; Wu, H.-T.; Chang, T.-C.; Jan, F.-D.; Lin, C.-C. Site-specific immobilization of enzymes on magnetic nanoparticles and their use in organic synthesis. Bioconjugate Chem. 2012, 23, 714–724. [Google Scholar] [CrossRef]
- Nahalka, J.; Liu, Z.; Chen, X.; Wang, P.G. Superbeads: Immobilization in “sweet” chemistry. Chem. Eur. J. 2003, 9, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.-Y.; Tsai, S.-W.; Chen, T.-L. Improvements of enzyme activity and enantioselectivity via combined substrate engineering and covalent immobilization. Biotechnol. Bioeng. 2008, 101, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ge, J.; Liu, Z. Enhanced activity of immobilized or chemically modified enzymes. ACS Catal. 2015, 5, 4503–4513. [Google Scholar] [CrossRef]
- Chibata, I.; Tosa, T.; Sato, T.; Mori, T. Production of L-amino acids by aminoacylase adsorbed on DEAE-sephadex. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1976; Volume 44, pp. 746–759. [Google Scholar]
- Petronijević, Ž.; Ristić, S.; Pešić, D.; Šmelcerović, A. Immobilization of dextransucrase on regenerated benzoyl cellulose carriers. Enzyme Microb. Technol. 2007, 40, 763–768. [Google Scholar] [CrossRef]
- Schöffer, J.d.N.; Klein, M.P.; Rodrigues, R.C.; Hertz, P.F. Continuous production of β-cyclodextrin from starch by highly stable cyclodextrin glycosyltransferase immobilized on chitosan. Carbohydr. Polym. 2013, 98, 1311–1316. [Google Scholar] [CrossRef]
- Schöffer, J.d.N.; Matte, C.R.; Charqueiro, D.S.; de Menezes, E.W.; Costa, T.M.H.; Benvenutti, E.V.; Rodrigues, R.C.; Hertz, P.F. Directed immobilization of CGTase: The effect of the enzyme orientation on the enzyme activity and its use in packed-bed reactor for continuous production of cyclodextrins. Process. Biochem. 2017, 58, 120–127. [Google Scholar] [CrossRef]
- Rakmai, J.; Cheirsilp, B.; Prasertsan, P. Enhanced thermal stability of cyclodextrin glycosyltransferase in alginate–gelatin mixed gel beads and the application for β-cyclodextrin production. Biocatal. Agric. Biotechnol. 2015, 4, 717–726. [Google Scholar] [CrossRef]
- Jung, D.-H.; Jung, J.-H.; Seo, D.-H.; Ha, S.-J.; Kweon, D.-K.; Park, C.-S. One-pot bioconversion of sucrose to trehalose using enzymatic sequential reactions in combined cross-linked enzyme aggregates. Bioresour. Technol. 2013, 130, 801–804. [Google Scholar] [CrossRef]
- Orrego, A.H.; Trobo-Maseda, L.; Rocha-Martin, J.; Guisan, J.M. Immobilization-stabilization of a complex multimeric sucrose synthase from Nitrosomonas europaea. Synthesis of UDP-glucose. Enzyme Microb. Technol. 2017, 105, 51–58. [Google Scholar] [CrossRef]
- De Winter, K.; Soetaert, W.; Desmet, T. An imprinted cross-linked enzyme aggregate (iCLEA) of sucrose phosphorylase: Combining improved stability with altered specificity. Int. J. Mol. Sci. 2012, 13, 11333. [Google Scholar] [CrossRef]
- Kaulpiboon, J.; Pongsawasdi, P.; Zimmermann, W. Molecular imprinting of cyclodextrin glycosyltransferases from Paenibacillus sp. A11 and Bacillus macerans with γ-cyclodextrin. FEBS J. 2007, 274, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, S.; Li, W.; Li, R.; Chen, S.; Ri, H.I.; Kim, T.M.; Kang, M.S.; Sun, L.; Sun, X.; et al. Improvement of trehalose production by immobilized trehalose synthase from Thermus thermophilus HB27. Molecules 2018, 23, 1087. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef]
- Liese, A.; Hilterhaus, L. Evaluation of immobilized enzymes for industrial applications. Chem. Soc. Rev. 2013, 42, 6236–6249. [Google Scholar] [CrossRef]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef]
- Rakmai, J.; Cheirsilp, B. Continuous production of β-cyclodextrin by cyclodextrin glycosyltransferase immobilized in mixed gel beads: Comparative study in continuous stirred tank reactor and packed bed reactor. Biochem. Eng. J. 2016, 105, 107–113. [Google Scholar] [CrossRef]
- Cho, Y.-J.; Park, O.-J.; Shin, H.-J. Immobilization of thermostable trehalose synthase for the production of trehalose. Enzyme Microb. Technol. 2006, 39, 108–113. [Google Scholar] [CrossRef]
- Szymańska, K.; Odrozek, K.; Zniszczoł, A.; Pudło, W.; Jarzębski, A.B. A novel hierarchically structured siliceous packing to boost the performance of rotating bed enzymatic reactors. Chem. Eng. J. 2017, 315, 18–24. [Google Scholar] [CrossRef]
- Cattaneo, G.; Rabuffetti, M.; Speranza, G.; Kupfer, T.; Peters, B.; Massolini, G.; Ubiali, D.; Calleri, E. Synthesis of adenine nucleosides by transglycosylation using two sequential nucleoside phosphorylase-based bioreactors with on-line reaction monitoring by using HPLC. ChemCatChem 2017, 9, 4614–4620. [Google Scholar] [CrossRef]
- Szymańska, K.; Odrozek, K.; Zniszczoł, A.; Torrelo, G.; Resch, V.; Hanefeld, U.; Jarzębski, A.B. MsAcT in siliceous monolithic microreactors enables quantitative ester synthesis in water. Catal. Sci. Technol. 2016, 6, 4882–4888. [Google Scholar] [CrossRef] [Green Version]
- Szymańska, K.; Pudło, W.; Mrowiec-Białoń, J.; Czardybon, A.; Kocurek, J.; Jarzębski, A.B. Immobilization of invertase on silica monoliths with hierarchical pore structure to obtain continuous flow enzymatic microreactors of high performance. Microporous Mesoporous Mater. 2013, 170, 75–82. [Google Scholar] [CrossRef]
- Lawrence, J.; O’Sullivan, B.; Lye, G.J.; Wohlgemuth, R.; Szita, N. Microfluidic multi-input reactor for biocatalytic synthesis using transketolase. J. Mol. Catal. B Enzym. 2013, 95, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
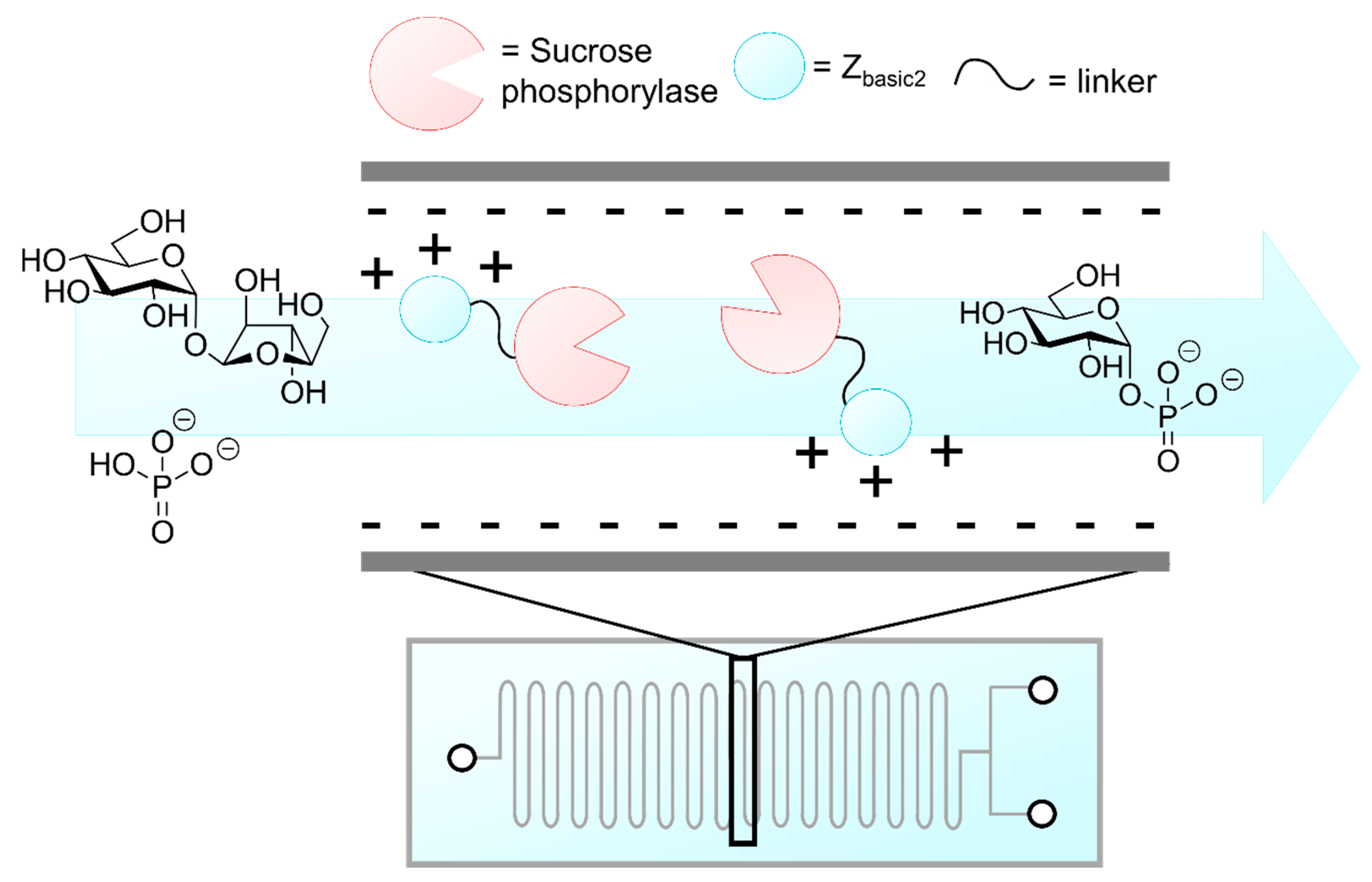
- Valikhani, D.; Bolivar, J.M.; Pfeiffer, M.; Nidetzky, B. Multivalency effects on the immobilization of sucrose phosphorylase in flow microchannels and their use in the development of a high-performance biocatalytic microreactor. ChemCatChem 2017, 9, 161–166. [Google Scholar] [CrossRef]
- Strub, D.J.; Szymańska, K.; Hrydziuszko, Z.; Bryjak, J.; Jarzębski, A.B. Continuous flow kinetic resolution of a non-equimolar mixture of diastereoisomeric alcohol using a structured monolithic enzymatic microreactor. React. Chem. Eng. 2019, 4, 587–594. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Woodley, J.M. Role of biocatalysis in sustainable chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef]
- Aurell, C.-J.; Karlsson, S.; Pontén, F.; Andersen, S.M. Lipase catalyzed regioselective lactamization as a key step in the synthesis of N-BOC-(2R)-1,4-oxazepane-2-carboxylic acid. Org. Process. Res. Develop. 2014, 18, 1116–1119. [Google Scholar] [CrossRef]
- Mallin, H.; Muschiol, J.; Byström, E.; Bornscheuer, U.T. Efficient biocatalysis with immobilized enzymes or encapsulated whole cell microorganism by using the spinchem reactor system. ChemCatChem 2013, 5, 3529–3532. [Google Scholar] [CrossRef]
- Carberry, J.J. Designing laboratory catalytic reactors. Ind. Eng. Chem. 1964, 56, 39–46. [Google Scholar] [CrossRef]
- Woodley, J.M.; Titchener-Hooker, N.J. The use of windows of operation as a bioprocess design tool. Bioprocess. Eng. 1996, 14, 263–268. [Google Scholar] [CrossRef]
- Wohlgemuth, R.; Plazl, I.; Žnidaršič-Plazl, P.; Gernaey, K.V.; Woodley, J.M. Microscale technology and biocatalytic processes: Opportunities and challenges for synthesis. Trends Biotechnol. 2015, 33, 302–314. [Google Scholar] [CrossRef]
- Rossetti, I. Continuous flow (micro-)reactors for heterogeneously catalyzed reactions: Main design and modelling issues. Catal. Today 2018, 308, 20–31. [Google Scholar] [CrossRef]

















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mestrom, L.; Przypis, M.; Kowalczykiewicz, D.; Pollender, A.; Kumpf, A.; Marsden, S.R.; Bento, I.; Jarzębski, A.B.; Szymańska, K.; Chruściel, A.; et al. Leloir Glycosyltransferases in Applied Biocatalysis: A Multidisciplinary Approach. Int. J. Mol. Sci. 2019, 20, 5263. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215263
Mestrom L, Przypis M, Kowalczykiewicz D, Pollender A, Kumpf A, Marsden SR, Bento I, Jarzębski AB, Szymańska K, Chruściel A, et al. Leloir Glycosyltransferases in Applied Biocatalysis: A Multidisciplinary Approach. International Journal of Molecular Sciences. 2019; 20(21):5263. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215263
Chicago/Turabian StyleMestrom, Luuk, Marta Przypis, Daria Kowalczykiewicz, André Pollender, Antje Kumpf, Stefan R. Marsden, Isabel Bento, Andrzej B. Jarzębski, Katarzyna Szymańska, Arkadiusz Chruściel, and et al. 2019. "Leloir Glycosyltransferases in Applied Biocatalysis: A Multidisciplinary Approach" International Journal of Molecular Sciences 20, no. 21: 5263. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215263