Peptidylarginine Deiminase Isozyme-Specific PAD2, PAD3 and PAD4 Inhibitors Differentially Modulate Extracellular Vesicle Signatures and Cell Invasion in Two Glioblastoma Multiforme Cell Lines

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
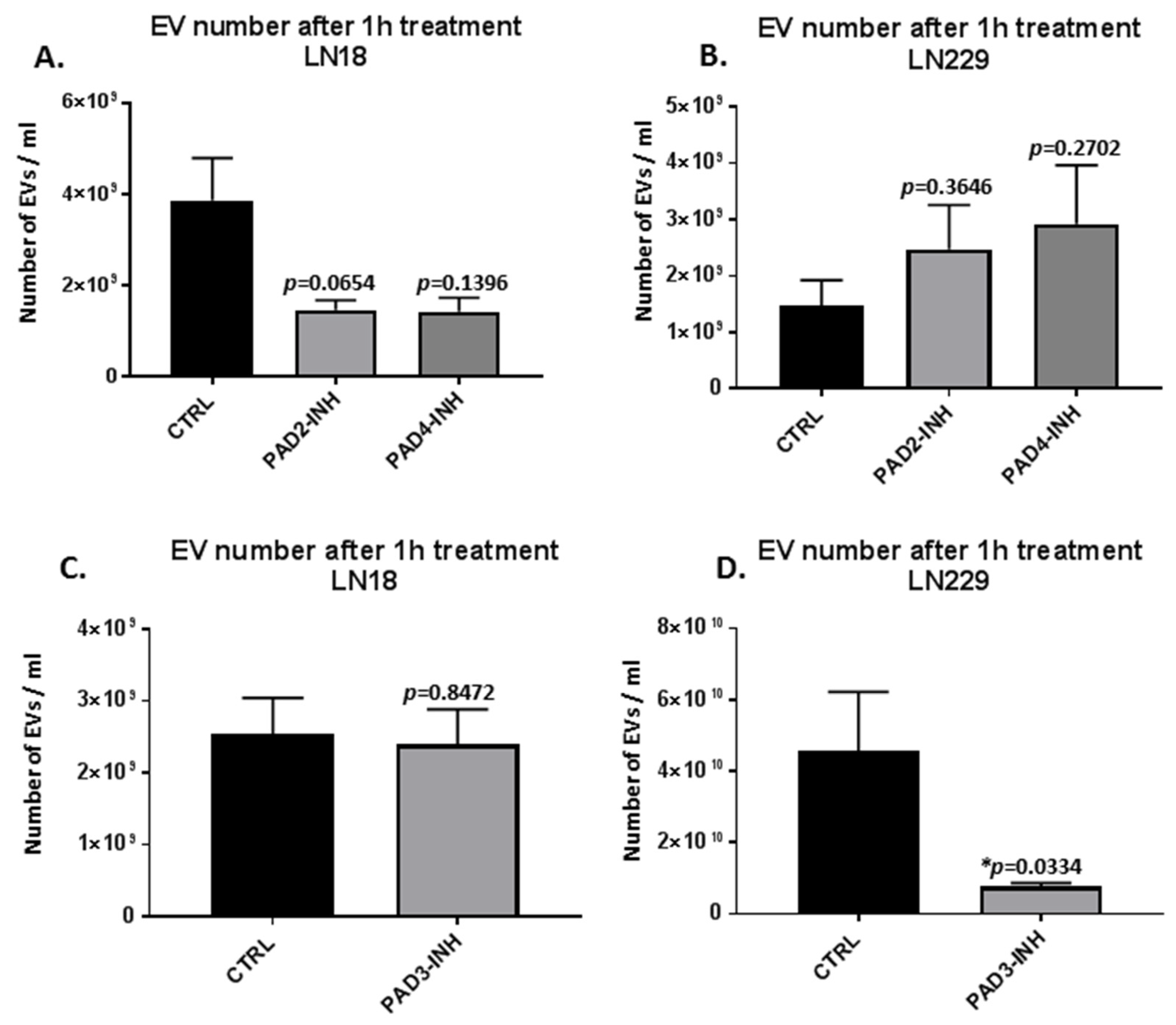
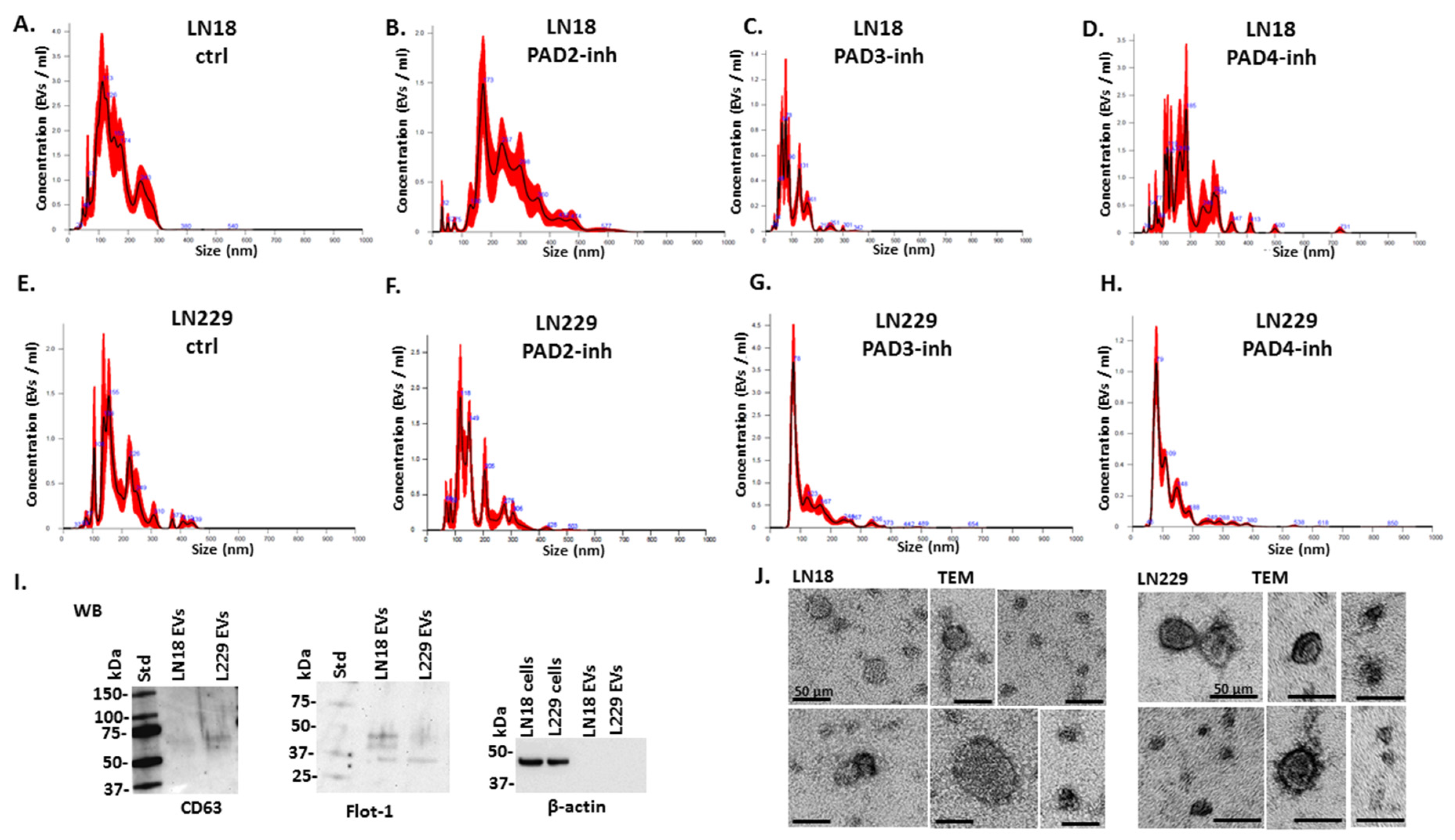
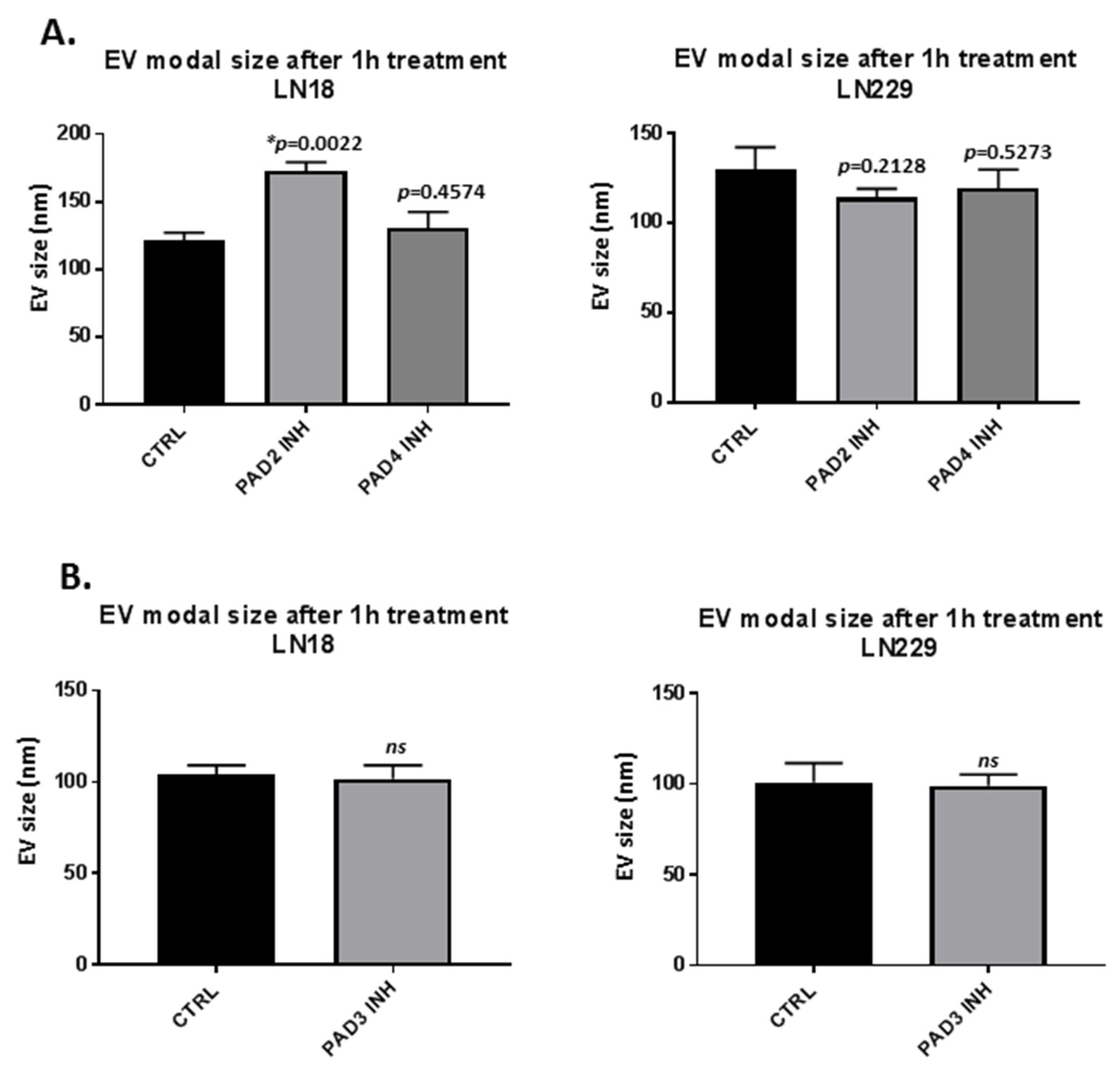
2.1. PAD Isozyme-Specific Inhibitors Differently Modulate EV Release in LN18 and LN229 GBM Cells Following 1 h Treatment
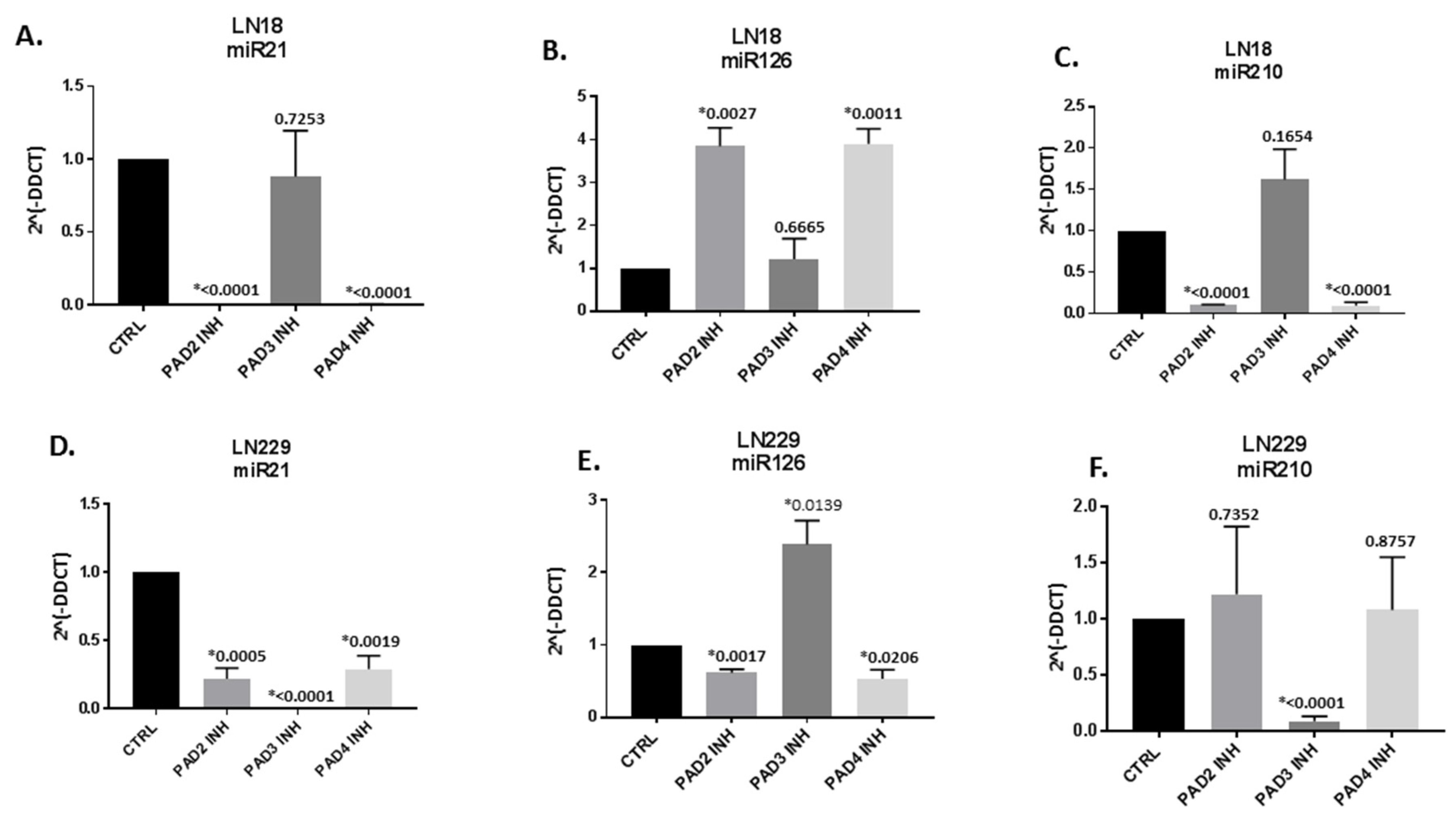
2.2. MicroRNA EV-cargo is Differently Modulated in Response to 1 h PAD Isozyme-Specific Inhibitor Treatment in LN18 and LN229 GBM Cells
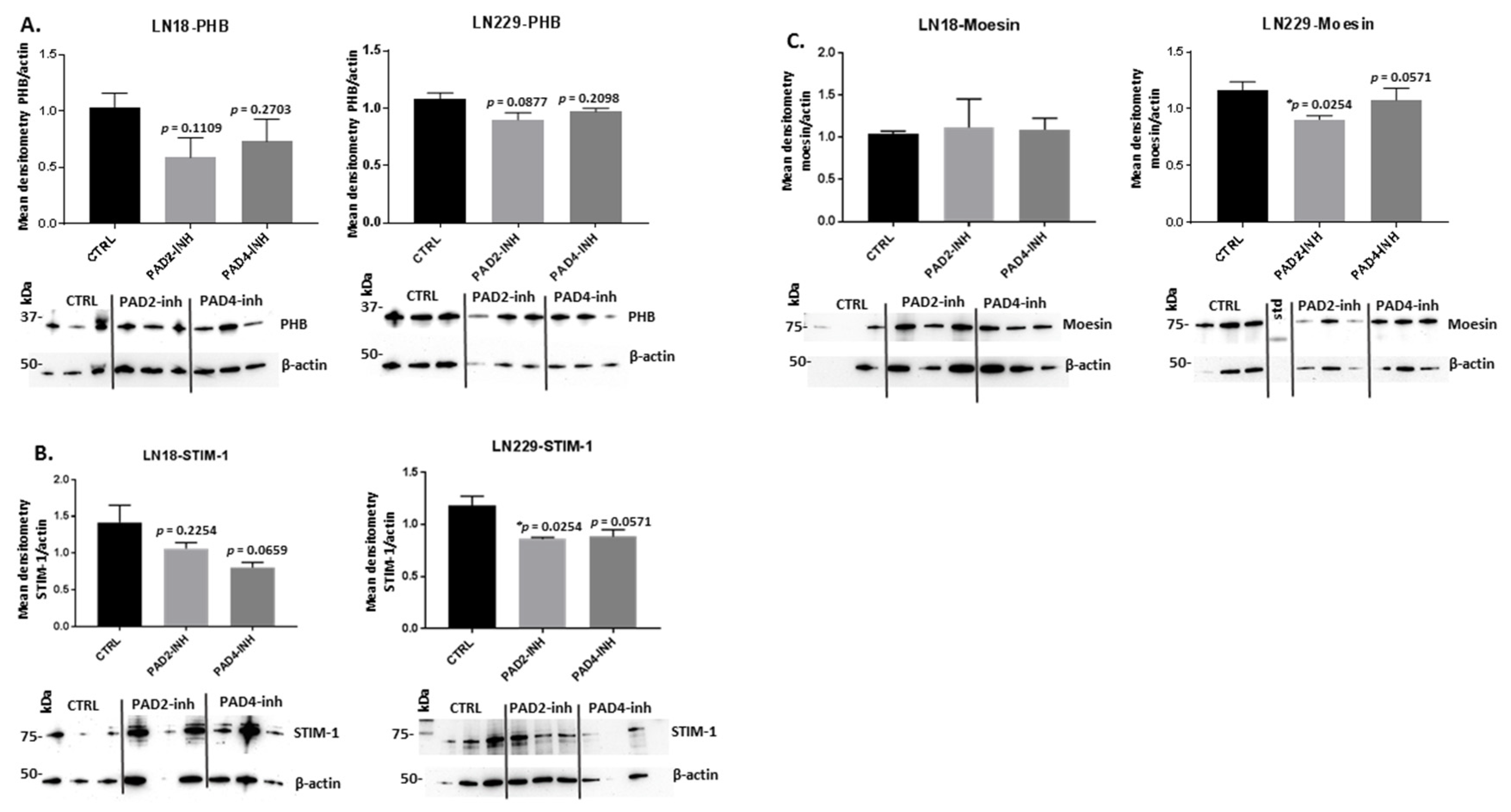
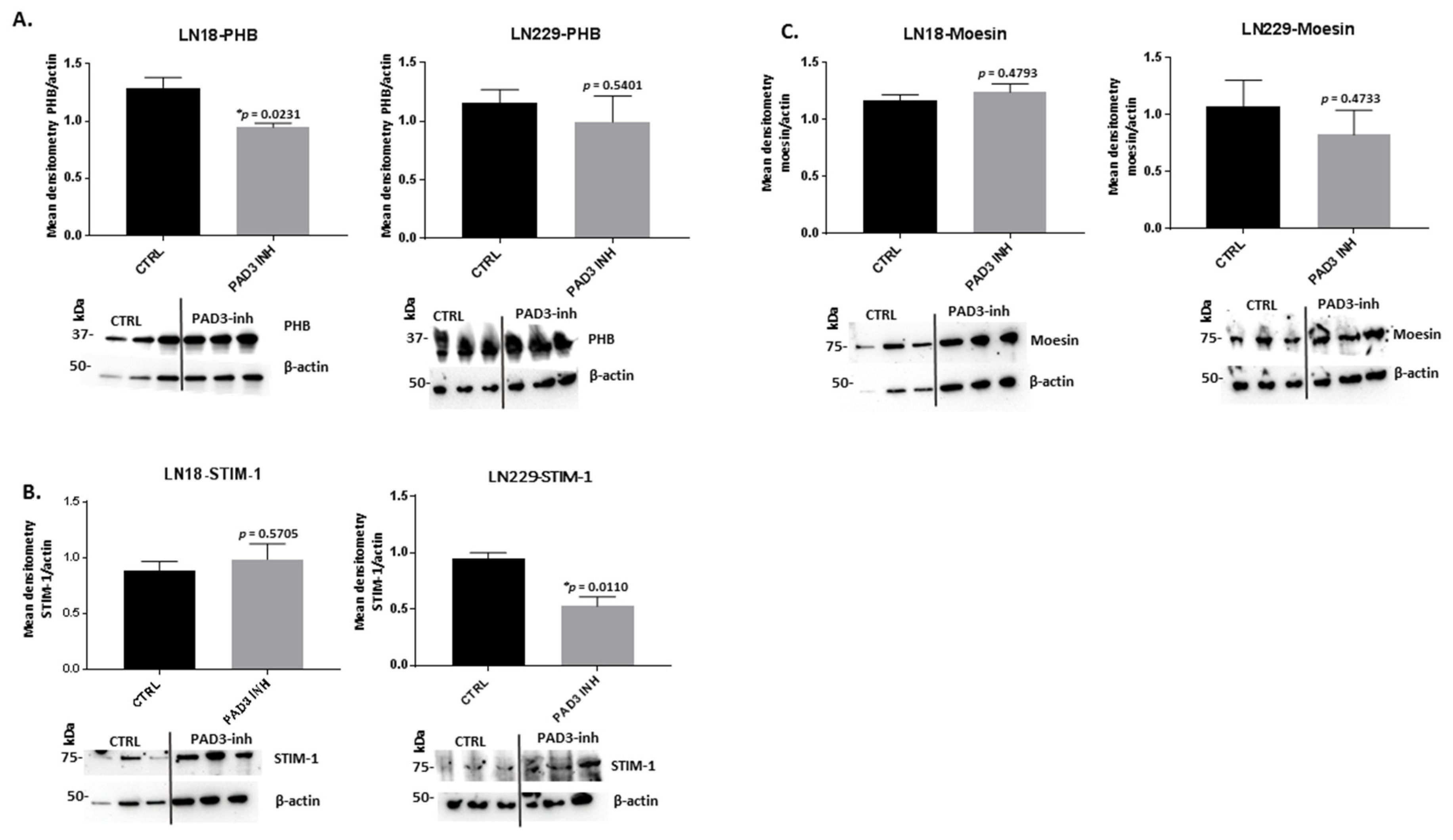
2.3. PAD Isozyme-Specific Inhibitors Affect PHB, STIM-1 and Moesin Protein Expression Differently in LN18 and LN229 GBM Cells Following 1 h Treatment
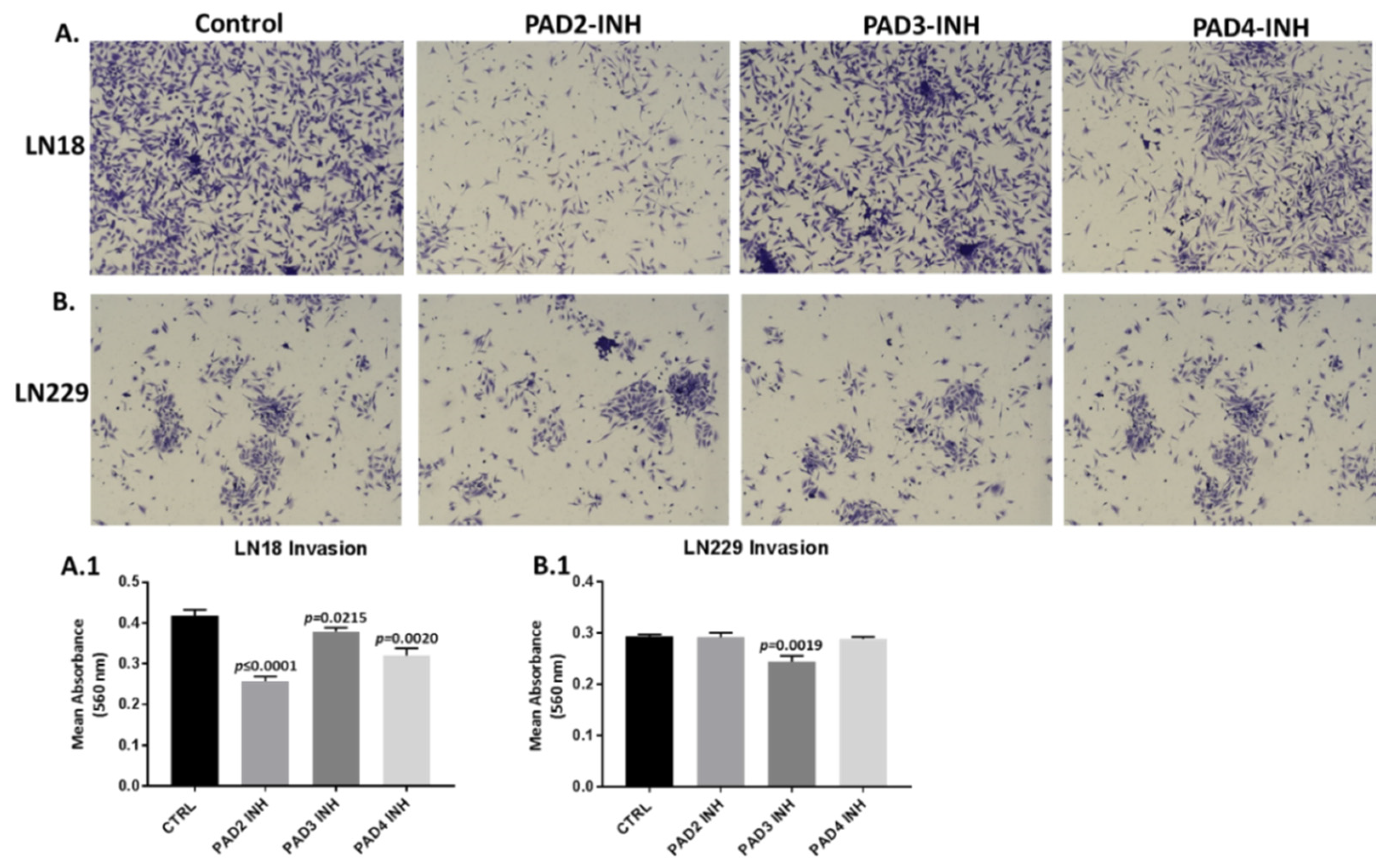
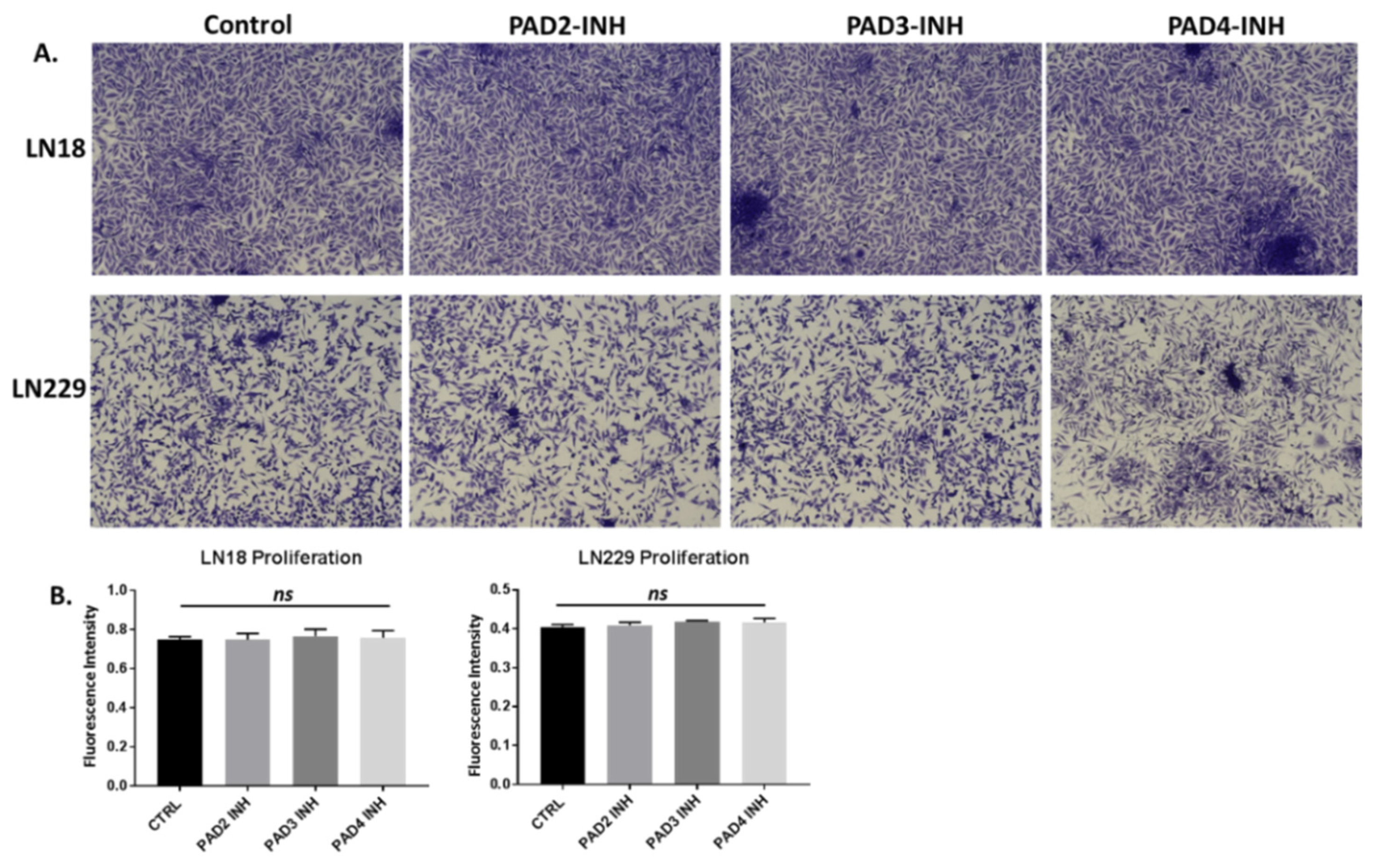
2.4. PAD Isozyme-Specific Inhibitors Differently Affect Invasion in LN18 and LN229 GBM Cells
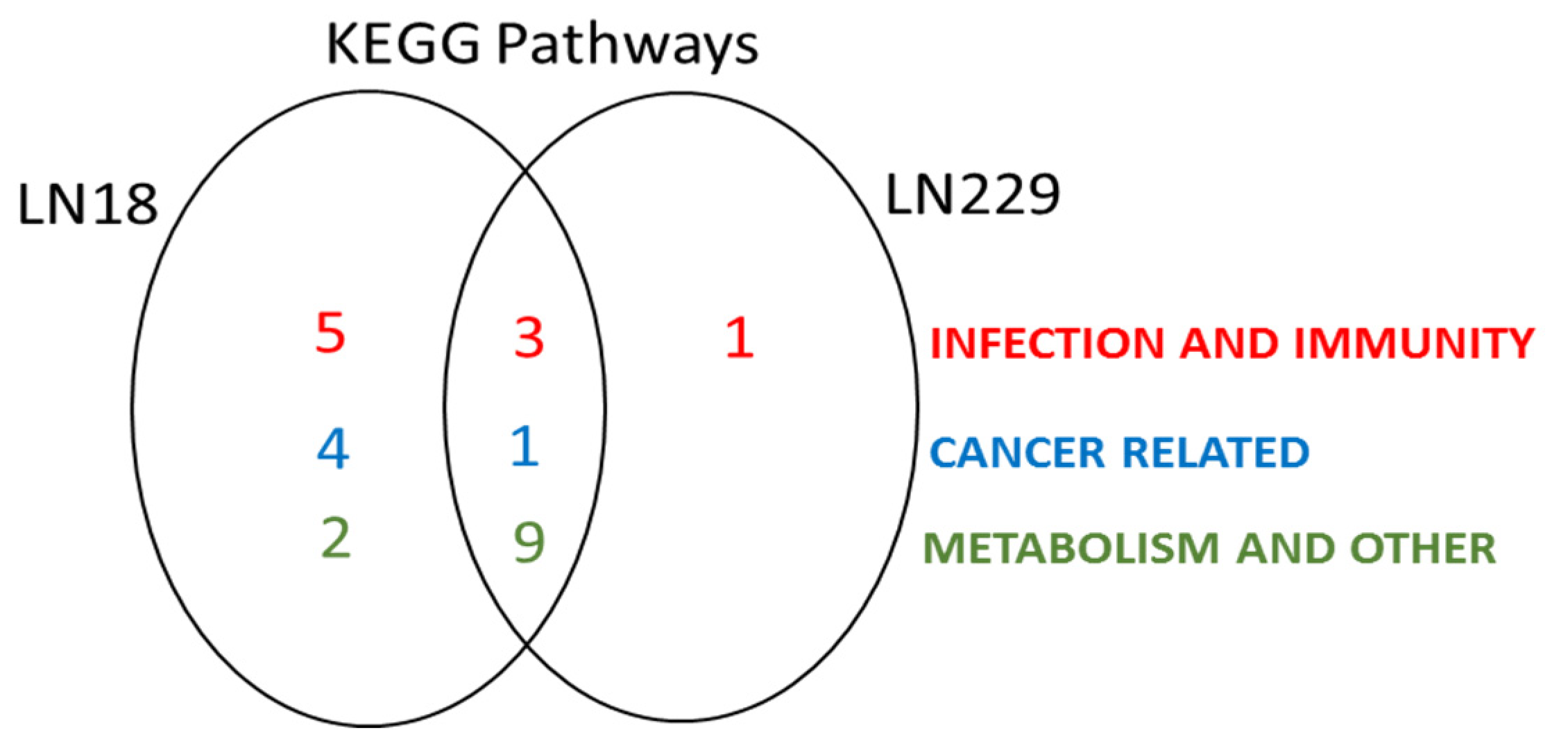
2.5. Deiminated Protein Targets and KEGG Networks Enriched in Deiminated Proteins Differ in LN18 and LN229 GBM Cells under Standard Culture Conditions
3. Discussion
4. Materials and Methods
4.1. GBM Cell Cultures and PAD-Inhibitor Treatment
4.2. Cell Viability Assays following PAD Inhibitor Treatment
4.3. Modulation of EV Release Using PAD2, PAD3 and PAD4 Isozyme-Specific Inhibitors Following 1 h Treatment
4.4. EV Isolation and Quantification by Nanoparticle Tracking Analysis
4.5. EV Characterisation by Transmission Electron Microscopy
4.6. Analysis of microRNAs miR21, miR126 and miR210 in GBM Cell EV-Cargo Following 1h PAD Inhibitor Treatment
4.7. Western Blotting Analysis
4.8. Cancer Cell Invasion Assay
4.9. Assessment of KEGG Pathways for Deiminated Proteins in LN18 and LN229 GBM Cells under Standard Culture Conditions
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | Bovine serum albumin |
| CD63 | CD63 antigen; granulophysin; lysosomal-associated membrane protein 3 |
| ECL | Enhanced chemiluminescence |
| EVs | Extracellular vesicles |
| F95 | Pan-deimination/citrullination antibody |
| FBS | Foetal bovine serum |
| GBM | Glioblastoma multiforme |
| Flot-1 | Flotillin-1 |
| HIF | Hypoxia-inducible factor |
| kDa | Kilodalton |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LC-MS/MS | Liquid chromatography mass spectrometry |
| miR | microRNA |
| NTA | Nanoparticle tracking analysis |
| PAD | Peptidylarginine deiminase |
| PHB | Prohibitin |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| TBS | Tris buffered saline |
| TEM | Transmission electron microscopy |
| STIM-1 | Stromal interaction molecule 1 |
| WB | Western blotting |
References
- Vossenaar, E.R.; Zendman, A.J.; van Venrooij, W.J.; Pruijn, G.J. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. Bioessays 2003, 25, 1106–1118. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim. Biophy. Acta. 2013, 1829, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Witalison, E.E.; Thompson, P.R.; Hofseth, L.J. Protein Arginine Deiminases and Associated Citrullination: Physiological Functions and Diseases Associated with Dysregulation. Curr. Drug Targets 2015, 16, 700–710. [Google Scholar] [CrossRef]
- Henderson, B.; Martin, A.C. Protein moonlighting: A new factor in biology and medicine. Biochem. Soc. Trans. 2014, 42, 1671–1678. [Google Scholar] [CrossRef]
- Jeffrey, C.J. Protein moonlighting: What is it, and why is it important? Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160523. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, S.; Cherrington, B.D.; Horibata, S.; McElwee, J.L.; Thompson, P.R.; Coonrod, S.A. Potential role of peptidylarginine deiminase enzymes and protein citrullination in cancer pathogenesis. Biochem. Res. Int. 2012, 2012, 895343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholas, A.P.; Lu, L.; Heaven, M.; Kadish, I.; van Groen, T.; Accavitti-Loper, M.A.; Wewering, S.; Kofskey, D.; Gambetti, P.; Brenner, M. Ongoing studies of deimination in neurodegenerative diseases using the F95 antibody. In Protein Deimination in Human Health and Disease; Nicholas, A.P., Bhattacharya, S.K., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 257–280. [Google Scholar]
- Lange, S.; Gallagher, M.; Kholia, S.; Kosgodage, U.S.; Hristova, M.; Hardy, J.; Inal, J.M. Peptidylarginine Deiminases–Roles in Cancer and Neurodegeneration and Possible Avenues for Therapeutic Intervention via Modulation of Exosome and Microvesicle (EMV) Release? Int. J. Mol. Sci. 2017, 18, 1196. [Google Scholar] [CrossRef] [PubMed]
- Kosgodage, U.S.; Uysal-Onganer, P.; MacLatchy, A.; Kraev, I.; Chatterton, N.P.; Nicholas, A.P.; Inal, J.M.; Lange, S. Peptidylarginine Deiminases Post-Translationally Deiminate Prohibitin and Modulate Extracellular Vesicle Release and MicroRNAs in Glioblastoma Multiforme. Int. J. Mol. Sci. 2018, 20, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Valeriani, M.; Osti, M.F.; De Paula, U.; Lanzetta, G.; Tombolini, V.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodbelt, A.; Greenberg, D.; Winters, T.; Williams, M.; Vernon, S.; Collins, V.P. UK National Cancer Information Network Brain Tumour Group. Glioblastoma in England: 2007–2011. Eur. J. Cancer 2015, 51, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, T.; Lachance, D.H.; Molinaro, A.M.; Eckel-Passow, J.E.; Walsh, K.M.; Barnholtz-Sloan, J.; Ostrom, Q.T.; Francis, S.S.; Wiemels, J.; Robert, B.; et al. Understanding inherited genetic risk of adult glioma—A review. Neurooncol. Pract. 2016, 3, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parney, I.F. Basic concepts in glioma immunology. Adv. Exp. Med. Biol. 2012, 746, 42–52. [Google Scholar] [PubMed]
- Rodrigues, J.C.; Gonzalez, G.C.; Zhang, L.; Ibrahim, G.; Kelly, J.J.; Gustafson, M.P.; Lin, Y.; Dietz, A.B.; Forsyth, P.A.; Yong, W.V.; et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. 2010, 12, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Inal, J.M.; Kosgodage, U.; Azam, S.; Stratton, D.; Antwi-Baffour, S.; Lange, S. Blood/plasma secretome and microvesicles. Biochim. Biophys. Acta 2013, 1834, 2317–2325. [Google Scholar] [CrossRef]
- Fatima, F.; Nawaz, M. Vesiculated Long Non-Coding RNAs: Offshore Packages Deciphering Trans-Regulation between Cells, Cancer Progression and Resistance to Therapies. Non Coding RNA 2017, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Barbagallo, D.; Caponnetto, A.; Cirnigliaro, M.; Brex, D.; Barbagallo, C.; D’Angeli, F.; Morrone, A.; Caltabiano, R.; Barbagallo, G.M.; Ragusa, M.; et al. CircSMARCA5 Inhibits Migration of Glioblastoma Multiforme Cells by Regulating a Molecular Axis Involving Splicing Factors SRSF1/SRSF3/PTB. Int. J. Mol. Sci. 2018, 19, 480. [Google Scholar] [CrossRef] [Green Version]
- de Mooij, T.; Peterson, T.E.; Evans, J.; McCutcheon, B.; Parney, I.F. Short non-coding RNA sequencing of glioblastoma extracellular vesicles. J. Neurooncol. 2020, 146, 253–263. [Google Scholar] [CrossRef]
- Müller Bark, J.; Kulasinghe, A.; Chua, B.; Day, B.W.; Punyadeera, C. Circulating biomarkers in patients with glioblastoma. Br. J. Cancer 2019, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, C.A.; Kaye, A.H.; Drummond, K.J.; Widodo, S.S.; Mantamadiotis, T.; Vella, L.J.; Stylli, S.S. Extracellular vesicles and their role in glioblastoma. Crit. Rev. Clin. Lab. Sci. 2019, 22, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.; Simon, T.; Vintu, M.; Solkin, B.; Koch, B.; Stewart, N.; Benstead-Hume, G.; Pearl, F.M.G.; Critchley, G.; Stebbing, J.; et al. Cell-derived extracellular vesicles can be used as a biomarker reservoir for glioblastoma tumor subtyping. Commun. Biol. 2019, 2, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André-Grégoire, G.; Bidère, N.; Gavard, J. Temozolomide affects Extracellular Vesicles Released by Glioblastoma Cells. Biochimie 2018, 155, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Peferoen, L.; Amor, S. Extracellular vesicles as modulators of cell-to-cell communication in the healthy and diseased brain. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130516. [Google Scholar] [CrossRef] [Green Version]
- Godlewski, J.; Krichevsky, A.M.; Johnson, M.D.; Chiocca, E.A.; Bronisz, A. Belonging to a network--microRNAs, extracellular vesicles, and the glioblastoma microenvironment. Neuro. Oncol. 2015, 17, 652–662. [Google Scholar] [CrossRef] [Green Version]
- Gourlay, J.; Morokoff, A.P.; Luwor, R.B.; Zhu, H.J.; Kaye, A.H.; Stylli, S.S. The emergent role of exosomes in glioma. J. Clin. Neurosci. 2017, 35, 13–23. [Google Scholar] [CrossRef]
- Anthiya, S.; Griveau, A.; Loussouarn, C.; Baril, P.; Garnett, M.; Issartel, J.P.; Garcion, E. MicroRNA-Based Drugs for Brain Tumors. Trends Cancer 2018, 4, 222–238. [Google Scholar] [CrossRef] [Green Version]
- Lefranc, F.; Le Rhun, E.; Kiss, R.; Weller, M. Glioblastoma quo vadis: Will migration and invasiveness reemerge as therapeutic targets? Cancer Treat. Rev. 2018, 68, 145–154. [Google Scholar] [CrossRef]
- Matarredona, E.R.; Pastor, A.M. Extracellular Vesicle-Mediated Communication between the Glioblastoma and Its Microenvironment. Cells 2019, 9, 96. [Google Scholar] [CrossRef] [Green Version]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D’Ilio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS ONE 2014, 9, e88193. [Google Scholar] [CrossRef] [Green Version]
- Jorfi, S.; Ansa-Addo, E.; Kholia, S.; Stratton, D.; Valley, S.; Lange, S.; Inal, J. Inhibition of microvesiculation sensitizes prostate cancer cells to chemotherapy and reduces docetaxel dose required to limit tumor growth in vivo. Sci. Rep. 2015, 5, 13006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kholia, S.; Jorfi, S.; Thompson, P.R.; Causey, C.P.; Nicholas, A.P.; Inal, J.; Lange, S. A Novel Role for Peptidylarginine Deiminases (PADs) in Microvesicle Release: A Therapeutic Potential for PAD Inhibitors to Sensitize Prostate Cancer Cells to Chemotherapy. J. Extracell. Vesicles 2015, 4, 26192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, R.; Aung, T.; Vogel, D.; Chapuy, B.; Wenzel, D.; Becker, S.; Sinzig, U.; Venkataramani, V.; von Mach, T.; Jacob, R.; et al. Nuclear Trapping through Inhibition of Exosomal Export by Indomethacin Increases Cytostatic Efficacy of Doxorubicin and Pixantrone. Clin. Cancer Res. 2016, 22, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidharan-Chari, V.; Kohan, H.G.; Asimakopoulos, A.G.; Sudha, T.; Sell, S.; Kannan, K.; Boroujerdi, M.; Davis, P.J.; Mousa, S.A. Microvesicle removal of anticancer drugs contributes to drug resistance in human pancreatic cancer cells. Oncotarget 2016, 7, 50365–50379. [Google Scholar] [CrossRef]
- Kosgodage, U.S.; Trindade, R.P.; Thompson, P.R.; Inal, J.M.; Lange, S. Chloramidine/Bisindolylmaleimide -I-Mediated Inhibition of Exosome and Microvesicle Release and Enhanced Efficacy of Cancer Chemotherapy. Int. J. Mol. Sci. 2017, 18, 1007. [Google Scholar] [CrossRef]
- Kosgodage, U.S.; Mould, R.; Henley, A.B.; Nunn, A.V.; Guy, G.W.; Thomas, E.L.; Inal, J.M.; Bell, J.D.; Lange, S. Cannabidiol (CBD) Is a Novel Inhibitor for Exosome and Microvesicle (EMV) Release in Cancer. Front. Pharmacol. 2018, 9, 889. [Google Scholar] [CrossRef] [Green Version]
- Kosgodage, U.S.; Uysal-Onganer, P.; MacLatchy, A.; Mould, R.; Nunn, A.V.; Guy, G.W.; Kraev, I.; Chatterton, N.P.; Thomas, E.L.; Inal, J.M.; et al. Cannabidiol Affects Extracellular Vesicle Release, miR21 and miR126, and Reduces Prohibitin Protein in Glioblastoma Multiforme Cells. Transl. Oncol. 2019, 12, 513–522. [Google Scholar] [CrossRef]
- Luo, Y.; Arita, K.; Bhatia, M.; Knuckley, B.; Lee, Y.H.; Stallcup, M.R.; Sato, M.; Thompson, P.R. Inhibitors and inactivators of protein arginine deiminase 4: Functional and structural characterization. Biochemistry 2006, 45, 11727–11736. [Google Scholar] [CrossRef] [Green Version]
- György, B.; Toth, E.; Tarcsa, E.; Falus, A.; Buzas, E.I. Citrullination: A posttranslational modification in health and disease. Int. J. Biochem. Cell Biol. 2006, 38, 1662–1677. [Google Scholar] [CrossRef]
- Peng, Y.T.; Chen, P.; Ouyang, R.Y.; Song, L. Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis 2015, 20, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Hernando-Rodríguez, B.; Artal-Sanz, M. Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin). Complex Cells 2018, 7, E238. [Google Scholar]
- Yang, J.; Li, B.; He, Q.Y. Significance of prohibitin domain family in tumorigenesis and its implication in cancer diagnosis and treatment. Cell Death Dis. 2018, 9, 580. [Google Scholar] [CrossRef] [PubMed]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathopulos, P.B.; Zheng, L.; Ikura, M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J. Biol. Chem. 2009, 284, 728–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflug. Arch. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Tang, Y.; Wang, F.; Zhang, H.; Xu, D.; Shen, Y.; Sun, S.; Yang, G. Blockade of store-operated Ca(2+) entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013, 330, 163–169. [Google Scholar] [CrossRef]
- Gallop, J.L. Filopodia and their links with membrane traffic and cell adhesion. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Wang, Q.; Lu, X.; Zhao, S.; Pang, M.; Wu, X.; Wu, H.; Hoffman, R.M.; Yang, Z.; Zhang, Y. Moesin Expression Is Associated with Glioblastoma Cell Proliferation and Invasion. Anticancer Res. 2017, 37, 2211–2218. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Lu, X.; Wang, J.; Yang, Z.; Hoffman, R.M.; Wu, X. Moesin Up-regulation Is Associated with Enhanced Tumor Progression Imaged Non-invasively in an Orthotopic Mouse Model of Human Glioblastoma. Anticancer Res. 2018, 38, 3267–3272. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Han, J. Expression of peptidylarginine deiminase type 4 (PAD4) in various tumors. Mol. Carcinog. 2006, 45, 183–196. [Google Scholar] [CrossRef]
- Masutomi, H.; Kawashima, S.; Kondo, Y.; Uchida, Y.; Jang, B.; Choi, E.K.; Kim, Y.S.; Shimokado, K.; Ishigami, A. Induction of peptidylarginine deiminase 2 and 3 by dibutyryl cAMP via cAMP-PKA signaling in human astrocytoma U-251MG cells. J. Neurosci. Res. 2017, 95, 1503–1512. [Google Scholar] [CrossRef]
- Sase, T.; Arito, M.; Onodera, H.; Omoteyama, K.; Kurokawa, M.S.; Kagami, Y.; Ishigami, A.; Tanaka, Y.; Kato, T. Hypoxia-induced production of peptidylarginine deiminases and citrullinated proteins in malignant glioma cells. Biochem. Biophys. Res. Commun. 2017, 482, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Rocha-Ferreira, E.; Thei, L.; Mawjee, P.; Bennett, K.; Thompson, P.R.; Subramanian, V.; Nicholas, A.P.; Peebles, D.; Hristova, M.; et al. Peptidylarginine deiminases: Novel drug targets for prevention of neuronal damage following hypoxic ischemic insult (HI) in neonates. J. Neurochem. 2014, 130, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Lange, S. Peptidylarginine Deiminases as Drug Targets in Neonatal Hypoxic-Ischemic Encephalopathy. Front. Neurol. 2016, 22, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamenter, M.E.; Uysal-Onganer, P.; Huynh, K.W.; Kraev, I.; Lange, S. Post-Translational Deimination of Immunological and Metabolic Protein Markers in Plasma and Extracellular Vesicles of Naked Mole-Rat (Heterocephalus glaber). Int. J. Mol. Sci. 2019, 20, 5378. [Google Scholar] [CrossRef] [Green Version]
- Magnadottir, B.; Uysal-Onganer, P.; Kraev, I.; Svansson, V.; Hayes, P.; Lange, S. Deiminated Proteins and Extracellular Vesicles-Novel Serum Biomarkers in Whales and Orca. Comp. Biochem. Physiol. D 2020, in press. [Google Scholar] [CrossRef]
- Subramanian, V.; Nicholas, A.P.; Thompson, P.R.; Ferretti, P. Modulation of calcium-induced cell death in human neural stem cells by the novel peptidylarginine deiminase-AIF pathway. Biochim. Biophys. Acta 2014, 1843, 1162–1171. [Google Scholar]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 2018, 5387041. [Google Scholar] [CrossRef]
- Hannen, R.; Selmansberger, M.; Hauswald, M.; Pagenstecher, A.; Nist, A.; Stiewe, T.; Acker, T.; Carl, B.; Nimsky, C.; Bartsch, J.W. Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-Like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor. Cancers 2019, 11, 921. [Google Scholar] [CrossRef] [Green Version]
- Barbano, R.; Palumbo, O.; Pasculli, B.; Galasso, M.; Volinia, S.; D’Angelo, V.; Icolaro, N.; Coco, M.; Dimitri, L.; Graziano, P.; et al. A miRNA signature for defining aggressive phenotype and prognosis in gliomas. PLoS ONE 2014, 9, e108950. [Google Scholar] [CrossRef] [Green Version]
- Lai, N.S.; Wu, D.G.; Fang, X.G.; Lin, Y.C.; Chen, S.S.; Li, Z.B.; Xu, S.S. Serum microRNA-210 as a potential noninvasive biomarker for the diagnosis and prognosis of glioma. Br. J. Cancer 2015, 112, 1241–1246. [Google Scholar] [CrossRef]
- Toraih, E.A.; El-Wazir, A.; Abdallah, H.Y.; Tantawy, M.A.; Fawzy, M.S. Deregulated MicroRNA Signature Following Glioblastoma Irradiation. Cancer Control 2019, 26. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, J.; Sun, G.; Meng, H.; Wang, J.; Guan, Y.; Yin, Y.; Zhao, Z.; Dong, X.; Yin, S.; et al. Glioblastoma extracellular vesicles induce the tumour-promoting transformation of neural stem cells. Cancer Lett. 2019, 466, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yekula, A.; Minciacchi, V.R.; Morello, M.; Shao, H.; Park, Y.; Zhang, X.; Muralidharan, K.; Freeman, M.R.; Weissleder, R.; Lee, H.; et al. Large and small extracellular vesicles released by glioma cells in vitro and in vivo. J. Extracell. Vesicles. 2019, 9, 1689784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akers, J.C.; Ramakrishnan, V.; Kim, R.; Phillips, S.; Kaimal, V.; Mao, Y.; Hua, W.; Yang, I.; Fu, C.C.; Nolan, J.; et al. miRNA contents of cerebrospinal fluid extracellular vesicles in glioblastoma patients. J. Neurooncol. 2015, 123, 205–216. [Google Scholar] [CrossRef] [Green Version]
- van der Vos, K.E.; Abels, E.R.; Zhang, X.; Lai, C.; Carrizosa, E.; Oakley, D.; Prabhakar, S.; Mardini, X.; Crommentuijn, M.H.W.; Skog, J.; et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. 2016, 18, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Batagov, A.O.; Schinelli, S.; Wang, J.; Wang, Y.; El Fatimy, R.; Rabinovsky, R.; Balaj, L.; Chen, C.C.; Hochberg, F.; et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 2017, 8, 1145. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef] [Green Version]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Yin, Y.; Ornell, K.J.; Beliveau, A.; Jain, A. Modulation of MicroRNAs 34a and 21 Affects Viability, Senescence, and Invasion in Glioblastoma Multiforme. J. Biomed. Nanotechnol. 2016, 12, 1782–1797. [Google Scholar] [CrossRef]
- Costa, P.M.; Cardoso, A.L.; Custódia, C.; Cunha, P.; Pereira de Almeida, L.; Pedroso de Lima, M.C. MiRNA-21 silencing mediated by tumor-targeted nanoparticles combined with sunitinib: A new multimodal gene therapy approach for glioblastoma. J. Control. Release 2015, 207, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Monfared, H.; Jahangard, Y.; Nikkhah, M.; Mirnajafi-Zadeh, J.; Mowla, S.J. Potential Therapeutic Effects of Exosomes Packed With a miR-21-Sponge Construct in a Rat Model of Glioblastoma. Front. Oncol. 2019, 9, 782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.T.; Zhang, X.Q.; Zhuang, J.T.; Chan, H.L.; Li, C.H.; Leung, G.K. MicroRNA-21 inhibition enhances in vitro chemosensitivity of temozolomide-resistant glioblastoma cells. Anticancer Res. 2012, 32, 2835–2841. [Google Scholar] [PubMed]
- Han, I.B.; Kim, M.; Lee, S.H.; Kim, J.K.; Kim, S.H.; Chang, J.H.; Teng, Y.D. Down-regulation of MicroRNA-126 in Glioblastoma and its Correlation with Patient Prognosis: A Pilot Study. Anticancer Res. 2016, 36, 6691–6697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ge, P.; Ma, C. MiR-126 Regulates the ERK Pathway via Targeting KRAS to Inhibit the Glioma Cell Proliferation and Invasion. Mol. Neurobiol. 2017, 54, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.A.; Sandalcioglu, I.E.; Karsak, M. Cannabinoids in Glioblastoma Therapy: New Applications for Old Drugs. Front. Mol. Neurosci. 2018, 11, 159. [Google Scholar] [CrossRef]
- López-Valero, I.; Saiz-Ladera, C.; Torres, S.; Hernández-Tiedra, S.; García-Taboada, E.; Rodríguez-Fornés, F.; Barba, M.; Dávila, D.; Salvador-Tormo, N.; Guzmán, M.; et al. Targeting Glioma Initiating Cells with A combined therapy of cannabinoids and temozolomide. Biochem. Pharmacol. 2018, 157, 266–274. [Google Scholar] [CrossRef]
- Bavelloni, A.; Ramazzotti, G.; Poli, A.; Piazzi, M.; Focaccia, E.; Blalock, W.; Faenza, I. MiRNA-210: A Current Overview. Anticancer Res. 2017, 37, 6511–6521. [Google Scholar]
- Voloboueva, L.A.; Sun, X.; Xu, L.; Ouyang, Y.-B.; Giffard, R.G. Distinct effects of miR-210 reduction on neurogenesis: Increased neuronal survival of inflammation but reduced proliferation associated with mitochondrial enhancement. J. Neurosci. 2017, 37, 3072–3084. [Google Scholar] [CrossRef] [Green Version]
- Fasanaro, P.; D’Alessandra, Y.; Di Stefano, V.; Melchionna, R.; Romani, S.; Pompilio, G.; Capogrossi, M.C.; Martelli, F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand ephrin-A3. J. Biol. Chem. 2008, 283, 15878–15883. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Chen, C.; Yang, D.; Liao, Q.; Luo, H.; Wang, X.; Zhou, F.; Yang, X.; Yang, J.; Zeng, C.; et al. Mesenchymal stem cells-derived extracellular vesicles, via miR-210, improve infarcted cardiac function by promotion of angiogenesis. Biochim. Biophys. Acta 2017, 1863, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Favaro, E.; Ramachandran, A.; McCormick, R.; Gee, H.; Blancher, C.; Crosby, M.; Devlin, C.; Blick, C.; Buffa, F.; Li, J.-L.; et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS ONE 2010, 5, e10345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Le, Q.-T.; Giaccia, A.J. MiR-210–micromanager of the hypoxia pathway. Trends Mol. Med. 2010, 16, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegal, T.; Charbit, H.; Paldor, I.; Zelikovitch, B.; Canello, T.; Benis, A.; Wong, M.L.; Morokoff, A.P.; Kaye, A.H.; Lavon, I. Dynamics of circulating hypoxia-mediated miRNAs and tumor response in patients with high-grade glioma treated with bevacizumab. J. Neurosurg. 2016, 125, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, T.; Thomassen, M.; Jensen, S.S.; Larsen, M.J.; Sørensen, K.P.; Hermansen, S.K.; Kruse, T.A.; Kristensen, B.W. Acute hypoxia induces upregulation of microRNA-210 expression in glioblastoma spheroids. CNS Oncol. 2015, 4, 25–35. [Google Scholar] [CrossRef]
- Tabibkhooei, A.; Izadpanahi, M.; Arab, A.; Zare-Mirzaei, A.; Minaeian, S.; Rostami, A.; Mohsenian, A. Profiling of novel circulating microRNAs as a non-invasive biomarker in diagnosis and follow-up of high and low-grade gliomas. Clin. Neurol. Neurosurg. 2019, 190, 105652. [Google Scholar] [CrossRef]
- Agrawal, R.; Pandey, P.; Jha, P.; Dwivedi, V.; Sarkar, C.; Kulshreshtha, R. Hypoxic signature of microRNAs in glioblastoma: Insights from small RNA deep sequencing. BMC Genom. 2014, 15, 686. [Google Scholar] [CrossRef] [Green Version]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, H.-K.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1905. [Google Scholar] [CrossRef] [Green Version]
- Bergmeier, W.; Weidinger, C.; Zee, I.; Feske, S. Emerging roles of store-operated Ca²⁺ entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels 2013, 7, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Hughes, J.D.; Rollins, S.; Chen, B.; Perkins, E. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp. Mol. Pathol. 2011, 91, 753–760. [Google Scholar] [CrossRef]
- Chen, Y.F.; Chiu, W.T.; Chen, Y.T.; Lin, P.Y.; Huang, H.J.; Chou, C.Y.; Chang, H.-C.; Tang, M.-J.; Shen, M.R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 10, 15225–15230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karvar, S.; Ansa-Addo, E.A.; Suda, J.; Singh, S.; Zhu, L.; Li, Z.; Rockey, D.C. Hepatology. Moesin, an ERM family member, regulates hepatic fibrosis. Hepatology 2019, 31078. [Google Scholar] [CrossRef] [PubMed]
- Jacquemet, G.; Baghirov, H.; Georgiadou, M.; Sihto, H.; Peuhu, E.; Cettour-Janet, P.; He, T.; Perälä, M.; Kronqvist, P.; Joensuu, H.; et al. L-type calcium channels regulate filopodia stability and cancer cell invasion downstream of integrin signalling. Nat. Commun. 2016, 7, 13297. [Google Scholar] [CrossRef] [Green Version]
- Jacquemet, G.; Hamidi, H.; Ivaska, J. Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr. Opin. Cell Biol. 2015, 36, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Jacquemet, G.; Stubb, A.; Saup, R.; Miihkinen, M.; Kremneva, E.; Hamidi, H.; Ivaska, J. Filopodome Mapping Identifies p130Cas as a Mechanosensitive Regulator of Filopodia Stability. Curr. Biol. 2019, 29, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Morales, F.C.; Agarwal, N.K.; Dogruluk, T.; Gagea, M.; Georgescu, M.M. Moesin is a glioma progression marker that induces proliferation and Wnt/β-catenin pathway activation via interaction with CD44. Cancer Res. 2013, 73, 1142–1155. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Dong, M.; Cong, K.; Chen, Y.; Ma, Z. Correlations of Moesin expression with the pathological stage, nerve infiltration, tumor location and pain severity in patients with pancreatic cancer. J. BUON 2019, 24, 1225–1232. [Google Scholar]
- Yu, L.; Zhao, L.; Wu, H.; Zhao, H.; Yu, Z.; He, M.; Jin, F.; Wei, M. Moesin is an independent prognostic marker for ER-positive breast cancer. Oncol. Lett. 2019, 17, 1921–1933. [Google Scholar] [CrossRef] [Green Version]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex©: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K. Specificity and mode of action of the muscle-type protein-arginine deiminase. Arch. Biochem. Biophys. 1992, 293, 362–369. [Google Scholar] [CrossRef]
- Tarcsa, E.; Marekov, L.N.; Mei, G.; Melino, G.; Lee, S.C.; Steinert, P.M. Protein unfolding by peptidylarginine deiminase. Substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J. Biol. Chem. 1996, 271, 30709–30716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Womeldorff, M.; Gillespie, D.; Jensen, R.L. Hypoxia-inducible factor-1 and associated upstream and downstream proteins in the pathophysiology and management of glioblastoma. Neurosurg. Focus 2014, 37, E8. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.A.; Asuthkar, S.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. Cancer stem cell molecular reprogramming of the Warburg effect in glioblastomas: A new target gleaned from an old concept. CNS Oncol. 2016, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.J.; Fu, X.L.; Guang, R.; To, S.T. Advances in the targeting of HIF-1α and future therapeutic strategies for glioblastoma multiforme (Review). Oncol. Rep. 2017, 37, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Xu, P.; Sowers, J.L.; Machuca, D.F.; Mirfattah, B.; Herring, J.; Tang, H.; Chen, Y.; Tian, B.; Brasier, A.R.; et al. Proteome Analysis of Hypoxic Glioblastoma Cells Reveals Sequential Metabolic Adaptation of One-Carbon Metabolic Pathways. Mol. Cell Proteom. 2017, 16, 1906–1921. [Google Scholar] [CrossRef] [Green Version]
- Han, J.E.; Lim, P.W.; Na, C.M.; Choi, Y.S.; Lee, J.Y.; Kim, Y.; Park, H.W.; Moon, H.E.; Heo, M.S.; Park, H.R.; et al. Inhibition of HIF1α and PDK Induces Cell Death of Glioblastoma Multiforme. Exp. Neurobiol. 2017, 26, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Nauman, P.; Bonicki, W.; Michalik, R.; Warzecha, A.; Czernicki, Z. The concentration of thyroid hormones and activities of iodothyronine deiodinases are altered in human brain gliomas. Folia Neuropathol. 2004, 42, 67–73. [Google Scholar]
- Nauman, P. Thyroid hormones in the central nervous system (CNS) and their effect on neoplasm formation, particularly on the development and course of glioblastoma multiforme-research hypothesis. Endokrynol. Pol. 2015, 66, 444–459. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, S.; Vranic, S.; Cyprian, F.S.; Al Moustafa, A.E. Epstein-Barr Virus in Gliomas: Cause, Association, or Artifact? Front. Oncol. 2018, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Limam, S.; Missaoui, N.; Mestiri, S.; Yacoubi, M.T.; Krifa, H.; Selmi, B.; Mokni, M. Epstein-Barr virus infection in gliomas. Curr. Res. Transl. Med. 2019, 67, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.G.; Peng, Y.; Koussougbo, K.S. Necroptosis: A novel therapeutic target for glioblastoma. Med. Hypotheses 2011, 76, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.M.; Lang, F.F.; Aggarwal, B.B.; Fuller, G.N.; Wildrick, D.M.; Sawaya, R. Necrosis and glioblastoma: A friend or a foe? A review and a hypothesis. Neurosurgery 2002, 51, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Melo-Lima, S.; Celeste Lopes, M.; Mollinedo, F. Necroptosis is associated with low procaspase-8 and active RIPK1 and -3 in human glioma cells. Oncoscience 2014, 1, 649–664. [Google Scholar] [CrossRef]
- Yi, G.Z.; Xiang, W.; Feng, W.Y.; Chen, Z.Y.; Li, Y.M.; Deng, S.Z.; Guo, M.L.; Zhao, L.; Sun, X.G.; He, M.Y.; et al. Identification of Key Candidate Proteins and Pathways Associated with Temozolomide Resistance in Glioblastoma Based on Subcellular Proteomics and Bioinformatical Analysis. Biorned Res. Int. 2018, 2018, 5238760. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Wei, B.; Wang, L.; Wang, L.; Kong, D.; Jin, Y.; Sun, Z. Analysis of gene expression profiles associated with glioma progression. Mol. Med. Rep. 2015, 12, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Chiasserini, D.; Davidescu, M.; Orvietani, P.L.; Susta, F.; Macchioni, L.; Petricciuolo, M.; Castigli, E.; Roberti, R.; Binaglia, L.; Corazzi, L. 3-Bromopyruvate treatment induces alterations of metabolic and stress-related pathways in glioblastoma cells. J. Proteom. 2017, 152, 329–338. [Google Scholar] [CrossRef]
- Ramão, A.; Gimenez, M.; Laure, H.J.; Izumi, C.; Vida, R.C.; Oba-Shinjo, S.; Marie, S.K.; Rosa, J.C. Changes in the expression of proteins associated with aerobic glycolysis and cell migration are involved in tumorigenic ability of two glioma cell lines. Proteome Sci. 2012, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Marin-Valencia, I.; Cho, S.K.; Rakheja, D.; Hatanpaa, K.J.; Kapur, P.; Mashimo, T.; Jindal, A.; Vemireddy, V.; Good, L.B.; Raisanen, J.; et al. Glucose metabolism via the pentose phosphate pathway, glycolysis and Krebs cycle in an orthotopic mouse model of human brain tumors. NMR Biomed. 2012, 25, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beinat, C.; Patel, C.B.; Xie, Y.; Gambhir, S.S. Evaluation of Glycolytic Response to Multiple Classes of Anti-glioblastoma Drugs by Noninvasive Measurement of Pyruvate Kinase M2 Using [18F]DASA-23. Mol. Imaging Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Subramanian, A.; Sarkar, R.R. Exploring the differences in metabolic behavior of astrocyte and glioblastoma: A flux balance analysis approach. Syst. Synth. Biol. 2015, 9, 159–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, J.L.; Kouri, F.M.; Hurley, L.A.; Liu, J.; Tommasini-Ghelfi, S.; Ji, Y.; Gao, P.; Calvert, A.E.; Lee, V.; Chandel, N.S.; et al. IDH3α regulates one-carbon metabolism in glioblastoma. Sci. Adv. 2019, 5, eaat0456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Immanuel, S.R.C.; Ghanate, A.D.; Parmar, D.S.; Marriage, F.; Panchagnula, V.; Day, P.J.; Raghunathan, A. Integrative analysis of rewired central metabolism in temozolomide resistant cells. Biochem. Biophys. Res. Commun. 2018, 495, 2010–2016. [Google Scholar] [CrossRef] [PubMed]
- Seluanov, A.; Gladyshev, V.N.; Vijg, J.; Gorbunova, V. Mechanisms of cancer resistance in long-lived mammals. Nat. Rev. Cancer. 2018, 18, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.S.; Asif, M.; Basheer, M.K.A.; Kang, C.W.; Al-Suede, F.S.; Ein, O.C.; Tang, J.; Majid, A.S.A.; Majid, A.M.S.A. Treatment of novel IL17A inhibitor in glioblastoma implementing 3rd generation co-culture cell line and patient-derived tumor model. Eur. J. Pharmacol. 2017, 803, 24–38. [Google Scholar] [CrossRef]
- Mehta, N.; Lyon, J.G.; Patil, K.; Mokarram, N.; Kim, C.; Bellamkonda, R.V. Bacterial Carriers for Glioblastoma Therapy. Mol. Ther. Oncolytics 2016, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Sait, M.; Rahmathulla, G.; Chen, T.L.; Barnett, G.H. Rare case of intracranial Salmonella enteritidis abscess following glioblastoma resection: Case report and review of the literature. Surg. Neurol. Int. 2011, 2, 149. [Google Scholar]
- Salmaggi, A.; Milanesi, I.; Silvani, A.; Gaviani, P.; Marchetti, M.; Fariselli, L.; Solero, C.L.; Maccagnano, C.; Casali, C.; Guzzetti, S.; et al. Prospective study of carmustine wafers in combination with 6-month metronomic temozolomide and radiation therapy in newly diagnosed glioblastoma: Preliminary results. J. Neurosurg. 2013, 118, 821–829. [Google Scholar] [CrossRef]
- Luciani, L.; Dubourg, G.; Graillon, T.; Honnorat, E.; Lepidi, H.; Drancourt, M.; Seng, P.; Stein, A. Salmonella enterica serovar Enteritidis brain abscess mimicking meningitis after surgery for glioblastoma multiforme: A case report and review of the literature. J. Med. Case Rep. 2016, 10, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhaddar, A.; Hall, W.; Boucetta, M. Subgaleal and brain abscesses due to Salmonella enteritidis following craniotomy for giant cell glioblastoma multiforme: A case report and literature review. Surg. Neurol. Int. 2019, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Kosgodage, U.S.; Matewele, P.; Mastroianni, G.; Kraev, I.; Brotherton, D.; Awamaria, B.; Nicholas, A.P.; Lange, S.; Inal, J.M. Peptidylarginine Deiminase Inhibitors Reduce Bacterial Membrane Vesicle Release and Sensitize Bacteria to Antibiotic Treatment. Front. Cell Infect. Microbiol. 2019, 9, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielecka, E.; Scavenius, C.; Kantyka, T.; Jusko, M.; Mizgalska, D.; Szmigielski, B.; Potempa, B.; Enghild, J.J.; Prossnitz, E.R.; Blom, A.M.; et al. Peptidyl arginine deiminase from Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J. Biol. Chem. 2014, 289, 32481–32487. [Google Scholar] [CrossRef] [Green Version]
- Muth, A.; Subramanian, V.; Beaumont, E.; Nagar, M.; Kerry, P.; McEwan, P.; Srinath, H.; Clancy, K.; Parelkar, S.; Thompson, P.R. Development of a Selective Inhibitor of Protein Arginine Deiminase 2. J. Med. Chem. 2017, 60, 3198–3211. [Google Scholar] [CrossRef] [Green Version]
- Knuckley, B.; Causey, C.P.; Jones, J.E.; Bhatia, M.; Dreyton, C.J.; Osborne, T.C.; Takahara, H.; Thompson, P.R. Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3. Biochemistry 2010, 49, 4852–4863. [Google Scholar] [CrossRef] [Green Version]
- Willis, V.C.; Banda, N.K.; Cordova, K.N.; Chandra, P.E.; Robinson, W.H.; Cooper, D.C.; Lugo, D.; Mehta, G.; Taylor, S.; Tak, P.P.; et al. Protein arginine deiminase 4 inhibition is sufficient for the amelioration of collagen-induced arthritis. Clin. Exp. Immunol. 2017, 188, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rasband, W.S.; Image, J.U.S. National Institutes of Health; Bethesda: Rockville, ML, USA, 2018. [Google Scholar]
- Uysal-Onganer, P.; Kawano, Y.; Caro, M.; Walker, M.M.; Diez, S.; Darrington, R.S.; Waxman, J.; Kypta, R.M. Wnt-11 promotes neuroendocrine-like differentiation, survival and migration of prostate cancer cells. Mol. Cancer 2010, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uysal-Onganer, P.; MacLatchy, A.; Mahmoud, R.; Kraev, I.; Thompson, P.R.; Inal, J.M.; Lange, S. Peptidylarginine Deiminase Isozyme-Specific PAD2, PAD3 and PAD4 Inhibitors Differentially Modulate Extracellular Vesicle Signatures and Cell Invasion in Two Glioblastoma Multiforme Cell Lines. Int. J. Mol. Sci. 2020, 21, 1495. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041495
Uysal-Onganer P, MacLatchy A, Mahmoud R, Kraev I, Thompson PR, Inal JM, Lange S. Peptidylarginine Deiminase Isozyme-Specific PAD2, PAD3 and PAD4 Inhibitors Differentially Modulate Extracellular Vesicle Signatures and Cell Invasion in Two Glioblastoma Multiforme Cell Lines. International Journal of Molecular Sciences. 2020; 21(4):1495. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041495
Chicago/Turabian StyleUysal-Onganer, Pinar, Amy MacLatchy, Rayan Mahmoud, Igor Kraev, Paul R. Thompson, Jameel M. Inal, and Sigrun Lange. 2020. "Peptidylarginine Deiminase Isozyme-Specific PAD2, PAD3 and PAD4 Inhibitors Differentially Modulate Extracellular Vesicle Signatures and Cell Invasion in Two Glioblastoma Multiforme Cell Lines" International Journal of Molecular Sciences 21, no. 4: 1495. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041495