Insights about MYC and Apoptosis in B-Lymphomagenesis: An Update from Murine Models

 , , , , ,
, , , , ,
Abstract
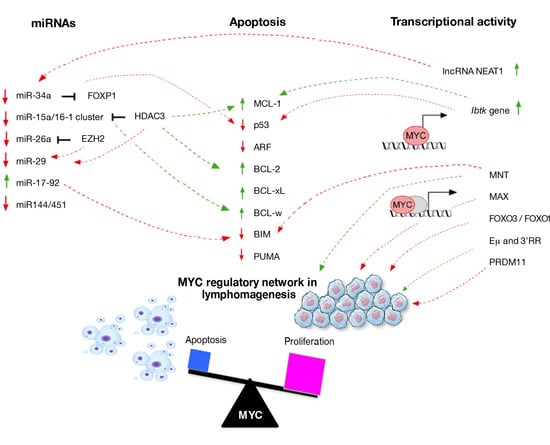
:
1. Role of MYC in Non-Transformed Cells
2. MYC and Lymphomagenesis
3. MYC Expression as a Key Factor of Apoptosis Activation
4. Crosstalk between MYC and Cellular Regulators of Apoptosis in Lymphoma
5. Transcriptional Regulation Network Involved in MYC-Driven Lymphomagenesis
6. Regulatory Feedback Action of miRs during MYC-Driven Lymphomagenesis
7. Conclusions and Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. MYC oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Eisenman, R.N. Deconstructing myc. Genes Dev. 2001, 15, 2023–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; Von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Choi, P.S.; Casey, S.C.; Dill, D.L.; Felsher, D.W. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell 2014, 26, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoepfler, P.S.; Zhang, X.Y.; Cheng, P.F.; Gafken, P.R.; McMahon, S.B.; Eisenman, R.N. Myc influences global chromatin structure. EMBO J. 2006, 25, 2723–2734. [Google Scholar] [CrossRef]
- Orian, A.; Steensel, B.; Delrow, J.; Bussemaker, H.J.; Li, L.; Sawado, T.; Williams, E.; Loo, L.W.M.; Cowley, S.M.; Yost, C.; et al. Genomic binding by the Drosophila myc, max, mad/mnt transcription factor network. Genes Dev. 2003, 17, 1101–1114. [Google Scholar] [CrossRef] [Green Version]
- Malynn, B.A.; De Alboran, I.M.; O’Hagan, R.C.; Bronson, R.; Davidson, L.; DePinho, R.A.; Alt, F.W. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000, 14, 1390–1399. [Google Scholar]
- Cohen, J.C.; Scott, D.K.; Miller, J.; Zhang, J.; Zhou, P.; Larson, J.E. Transient in utero knockout (TIUKO) of C-MYC affects late lung and intestinal development in the mouse. BMC Dev. Biol. 2004, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, K.; Cochran, B.H.; Stiles, C.D.; Leder, P. Cell-specific regulation of the c- myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell 1983, 35, 603–610. [Google Scholar] [CrossRef]
- Sears, R.; Leone, G.; DeGregori, J.; Nevins, J.R. Ras enhances Myc protein stability. Mol. Cell 1999, 3, 169–179. [Google Scholar]
- Kress, T.R.; Sabo, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb Perspect Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Allen-Petersen, B.L.; Sears, R.C. Mission Possible: Advances in MYC Therapeutic Targeting in Cancer. BioDrugs 2019, 33, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuyama, H.; Endo, H.; Akashika, T.; Kato, K.; Inoue, M. Downregulation of c-MYC protein levels contributes to cancer cell survival under dual deficiency of oxygen and glucose. Cancer Res. 2010, 70, 10213–10223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dani, C.; Blanchard, J.M.; Piechaczyk, M.; El Sabouty, S.; Marty, L.; Jeanteur, P. Extreme instability of myc mRNA in normal and transformed human cells. Proc. Natl. Acad. Sci USA 1984, 81, 7046–7050. [Google Scholar] [CrossRef] [Green Version]
- Wasylishen, A.R.; Penn, L.Z. Myc: The beauty and the beast. Genes Cancer 2010, 1, 532–541. [Google Scholar] [CrossRef] [Green Version]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [Green Version]
- Karube, K.; Campo, E. MYC alterations in diffuse large B-cell lymphomas. Semin. Hematol. 2015, 52, e106. [Google Scholar] [CrossRef]
- Cai, Q.; Medeiros, L.J.; Xiaolu, X.; Yuong, K.H. MYC-driven aggressive B-cell lymphomas: Biology, entity, differential diagnosis and clinical management. Oncotarget 2015, 6, 38591–38616. [Google Scholar] [CrossRef] [Green Version]
- Haralambieva, E.; Boerma, E.-J.; Van Imhoff, G.W.; Rosati, S.; Schuuring, E.; Müller-Hermelink, H.K.; Kluin, P.M.; Ott, G. Clinical, immunophenotypic, and genetic analysis of adult lymphomas with morphologic features of Burkitt lymphoma. Am. J. Surg. Pathol. 2005, 29, 1086–1094. [Google Scholar] [PubMed]
- Niitsu, N.; Okamoto, M.; Miura, I.; Hirano, M. Clinical features and prognosis of de novo diffuse large B-cell lymphoma with t (14; 18) and 8q24/c-MYC translocations. Leukemia 2009, 23, 777–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valera, A.; Balagué, O.; Colomo, L.; Martínez, A.; Delabie, J.; Taddesse-Heath, L.; Jaffe, E.S.; Campo, E. IG/MYC rearrangements are the main cytogenetic alteration in plasmablastic lymphomas. Am. J. Surg. Pathol. 2010, 34, 1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasylishen, A.R.; Chan-Seng-Yue, M.; Bros, C.; Dingar, D.; Tu, W.B.; Kalkat, M.; Chan, P.-K.; Mullen, P.J.; Huang, L.; Meyer, N.; et al. MYC phosphorylation at novel regulatory regions suppresses transforming activity. Cancer Res. 2013, 73, 6504–6515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuttler, F.; Ame, P.; Clark, H.; Haughey, C.; Mougin, C.; Cahn, J.Y.; Dang, C.V.; Raffeld, M.; Fest, T. C-myc box II mutations in Burkitt’s lymphoma-derived alleles reduce cell-transformation activity and lower response to broad apoptotic stimuli. Oncogenet 2001, 20, 6084–6094. [Google Scholar] [CrossRef] [Green Version]
- Herbst, A.; Hemann, M.T.; Tworkowski, K.A.; Salghetti, S.E.; Lowe, S.W.; Tansey, W.P. A conserved element in Myc that negatively regulates its proapoptotic activity. EMBO Rep. 2005, 6, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [Green Version]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef]
- Lefebure, M.; Tothill, R.W.; Kruse, E.; Hawkins, E.D.; Shortt, J.; Matthews, G.M.; Gregory, G.P.; Martin, B.P.; Kelly, M.J.; Todorovski, I.; et al. Genomic characterisation of Emu-Myc mouse lymphomas identifies Bcor as a Myc co-operative tumour-suppressor gene. Nat. Commun. 2017, 8, 14581. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, R.J.; Tumang, J.R.; Sinha, A.; Currier, N.; Cardiff, R.D.; Rothstein, T.L.; Faller, D.V.; Denis, G.V.E. mu-BRD2 transgenic mice develop B-cell lymphoma and leukemia. Blood 2004, 103, 1475–1484. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Jiang, C.; Zhang, Y.; Govande, A.; Trudeau, S.J.; Chen, F.; Fry, C.J.; Wolinsky, E.; Schineller, M.; Frost, T.C. Myc controls the Epstein-Barr virus lytic switch. Mol. Cell. 2020, 78, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Poe, J.C.; Minard-Colin, V.; Kountikov, E.I.; Haas, K.M.; Tedder, T.F. A c-Myc and surface CD19 signaling amplification loop promotes B cell lymphoma development and progression in mice. J. Immunol. 2012, 189, 2318–2325. [Google Scholar] [CrossRef] [Green Version]
- Vecchio, E.; Fiume, G.; Mignogna, C.; Iaccino, E.; Mimmi, S.; Maisano, D.; Trapasso, F.; Quinto, I. IBTK Haploinsufficiency Affects the Tumor Microenvironment of Myc-Driven Lymphoma in E-myc Mice. Int. J. Mol. Sci. 2020, 21, 885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlstrom, T.; Henriksson, M.A. Impact of MYC in regulation of tumor cell metabolism. Biochim. Biophys. Acta. 2015, 1849, 563–569. [Google Scholar] [CrossRef]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanità, G.; Ruocco, M.S.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Baylot, V.; Felsher, D.W. The MYC oncogene is a global regulator of the immune response. Blood 2018, 131, 2007–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsaves, V.; Tsesmetzi, N.; Chioureas, D.; Kis, L.; Leventaki, V.; Drakos, E.; Panaretakis, T.; Grander, D.; Medeiros, L.J.; Young, K.H.; et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia 2017, 31, 1633–1637. [Google Scholar] [CrossRef]
- Braun, J.; Felsher, D.W.; Goodglick, L.A. c-myc, MHCI, and NK resistance in immunodeficiency lymphomas. Ann. NY Acad. Sci. 1992, 651, 467–469. [Google Scholar] [CrossRef]
- Bisso, A.; Sabò, A.; Amati, B. MYC in Germinal Center-derived lymphomas: Mechanisms and therapeutic opportunities. Immunol. Rev. 2019, 288, 178–197. [Google Scholar] [CrossRef]
- Meyer, N.; Kim, S.S.; Penn, L.Z. The Oscar-worthy role of Myc in apoptosis. Semin. Cancer Biol. 2006, 16, 275–287. [Google Scholar] [CrossRef]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef] [PubMed]
- Dansen, T.B.; Whitfield, J.; Rostker, F.; Brown-Swigart, L.; Evan, G.I. Specific requirement for Bax, not Bak, in Myc-induced apoptosis and tumor suppression in vivo. J. Biol. Chem. 2006, 281, 10890–10895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eischen, C.M.; Woo, D.; Roussel, M.F.; Cleveland, J.L. Apoptosis triggered by Myc-induced suppression of Bcl-XL or Bcl-2 is bypassed during lymphomagenesis. Mol. Cell. Biol. 2001, 21, 5063–5070. [Google Scholar] [CrossRef] [Green Version]
- Juin, P.; Hunt, A.; Littlewood, T.; Griffiths, B.; Swigart, L.B.; Korsmeyer, S.; Evan, G. c-Myc functionally cooperates with Bax to induce apoptosis. Mol. Cell. Biol. 2002, 22, 6158–6169. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, Y.; Yunis, J.; Onorato-Showe, L.; Erikson, J.; Nowell, P.C.; Croce, C.M. Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science 1984, 224, 1403–1406. [Google Scholar] [CrossRef]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- Dunleavy, K. Double-hit lymphomas: Current paradigms and novel treatment approaches. Hematology Am. Soc. Hematol. Educ. Program. 2014, 2014, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, C.M.; Kim, A.S.; Mitra, R.; Choi, J.K.; Gong, J.Z.; Eischen, C.M. BCL-W has a fundamental role in B cell survival and lymphomagenesis. J. Clin. Invest. 2017, 127, 635–650. [Google Scholar] [CrossRef] [Green Version]
- Grabow, S.; Delbridge, A.R.; Aubrey, B.J.; Vandenberg, C.J.; Strasser, A. Loss of a Single Mcl-1 Allele Inhibits MYC-Driven Lymphomagenesis by Sensitizing Pro-B Cells to Apoptosis. Cell. Rep. 2016, 14, 2337–2347. [Google Scholar] [CrossRef] [Green Version]
- Grabow, S.; Kelly, G.L.; Delbridge, A.R.; Kelly, P.N.; Bouillet, P.; Adams, J.M.; Strasser, A. Critical B-lymphoid cell intrinsic role of endogenous MCL-1 in c-MYC-induced lymphomagenesis. Cell. Death Dis. 2016, 10, e2132. [Google Scholar] [CrossRef]
- Yu, L.; Yu, T.T.; Young, K.H. Cross-talk between Myc and p53 in B-cell lymphomas. Chronic Dis. Transl. Med. 2019, 22, 139–154. [Google Scholar]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch, A.; Prescott, J.; Shchors, K.; Hunt, A.; Soucek, L.; Dansen, T.B.; Swigart, L.B.; Evan, G.I. Bcl-xL gain of function and p19 ARF loss of function cooperate oncogenically with Myc in vivo by distinct mechanisms. Cancer Cell 2006, 10, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF–Mdm2–p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, C.A.; McCurrach, M.E.; De Stanchina, E.; Wallace-Brodeur, R.R.; Lowe, S.W. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 1999, 13, 2670–2677. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, C.; Lee, S.; Paulus-Hock, V.; Loddenkemper, C.; Eilers, M.; Schmitt, C.A. FoxO transcription factors suppress Myc-driven lymphomagenesis via direct activation of Arf. Genes Dev. 2007, 21, 2775–2787. [Google Scholar] [CrossRef] [Green Version]
- Alt, J.R.; Greiner, T.C.; Cleveland, J.L.; Eischen, C.M. Mdm2 haplo-insufficiency profoundly inhibits Myc-induced lymphomagenesis. EMBO J. 2003, 22, 1442–1450. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.J.; Scheijen, B.; Voncken, J.W.; Kieboom, K.; Berns, A.; Van Lohuizen, M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999, 13, 2678–2690. [Google Scholar] [CrossRef]
- Dickins, R.A.; Hemann, M.T.; Zilfou, J.T.; Simpson, D.R.; Ibarra, I.; Hannon, G.J.; Lowe, S.W. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nature Genet. 2005, 37, 1289–1295. [Google Scholar] [CrossRef]
- Haupt, Y.; Bath, M.L.; Harris, A.W.; Adams, J. Bmi-1 trans-gene induces lymphomas and collaborates with myc in tumourigenesis. Oncogenet 1993, 8, 3161–3164. [Google Scholar]
- Garrison, S.P.; Jeffers, J.R.; Yang, C.; Nilsson, J.A.; Hall, M.A.; Rehg, J.E.; Yue, W.; Yu, J.; Zhang, L.; Onciu, M.; et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol. Cell. Biol. 2008, 28, 5391–5402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; Van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef]
- Mathsyaraja, H.; Freie, B.; Cheng, P.F.; Babaeva, E.; Catchpole, J.T.; Janssens, D.; Henikoff, S.; Eisenman, R.N. Max deletion destabilizes MYC protein and abrogates Eµ-Myc lymphomagenesis. Genes Dev. 2019, 33, 1252–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Hurlin, P.J. MNT and emerging concepts of MNT-MYC antagonism. Genes 2017, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Vandenberg, C.J.; Anstee, N.S.; Hurlin, P.J.; Cory, S. Mnt modulates Myc-driven lymphomagenesis. Cell Death Differ. 2017, 24, 2117–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.V.; Vandenberg, C.J.; Ng, A.P.; Robati, M.R.; Anstee, N.S.; Rimes, J.; Hawkins, E.D.; Cory, S. Development and survival of MYC-driven lymphomas require the MYC antagonist MNT to curb MYC-induced apoptosis. Blood 2020, 135, 1019–1031. [Google Scholar] [CrossRef]
- Fog, C.K.; Asmar, F.; Côme, C.; Jensen, K.T.; Johansen, J.V.; Kheir, T.B.; Jacobsen, L.; Friis, C.; Louw, A.; Rosgaard, L.; et al. Loss of PRDM11 promotes MYC-driven lymphomagenesis. Blood 2015, 125, 1272–1281. [Google Scholar] [CrossRef] [Green Version]
- Vandenberg, C.J.; Motoyama, N.; Cory, S. FoxO3 suppresses Myc-driven lymphomagenesis. Cell Death Dis. 2016, 14, e2046. [Google Scholar] [CrossRef] [Green Version]
- Kabrani, E.; Chu, V.T.; Tasouri, E.; Sommermann, T.; Baßler, K.; Ulas, T.; Zenz, T.; Bullinger, L.; Schultze, J.L.; Rajewsky, K.; et al. Nuclear FOXO1 promotes lymphomagenesis in germinal center B cells. Blood 2018, 132, 2670–2683. [Google Scholar] [CrossRef] [Green Version]
- Dejean, A.S.; Hedrick, S.M.; Kerdiles, Y.M. Highly specialized role of Forkhead box O transcription factors in the immune system. Antioxid Redox Signal. 2011, 14, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlot, T.; Alt, F.W.; Bassing, C.H.; Suh, H.; Pinaud, E. Elucidation of IgH intronic enhancer functions via germ-line deletion. Proc Natl Acad Sci USA 2005, 102, 14362–14367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquet, M.; Garot, A.; Bender, S.; Carrion, C.; Rouaud, P.; Lecardeur, S.; Denizot, Y.; Cogné, M.; Pinaud, E. The Em enhancer region influences H chain expression and B cell fate without impacting IgVH repertoire and immune response in vivo. J. Immunol. 2014, 193, 1171–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saintamand, A.; Rouaud, P.; Garot, A.; Rios, G.; Cogné, M.; Denizot, Y. The IgH 39 regulatory region governs m chain transcription in mature B lymphocytes and the B cell fate. Oncotarget 2015, 6, 4845–4852. [Google Scholar] [CrossRef] [Green Version]
- Rouaud, P.; Vincent-Fabert, C.; Saintamand, A.; Fiancette, R.; Marquet, M.; Robert, I.; Reina-San-Martin, B.; Pinaud, E.; Cogné, M. The IgH 39 regulatory region controls somatic hypermutation in germinal center B cells. J. Exp. Med. 2013, 210, 1501–1507. [Google Scholar] [CrossRef] [Green Version]
- Saintamand, A.; Rouaud, P.; Saad, F.; Rios, G.; Cogne, Ã.Å.M.; Denizot, Y. Elucidation of IgH 39 region regulatory role during class switch recombination via germline deletion. Nat. Commun. 2015, 6, 7084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouaud, P.; Saintamand, A.; Saad, F.; Carrion, C.; Lecardeur, S.; Cogné, M.; Denizot, Y. Elucidation of the enigmatic IgD class-switch recombination via germline deletion of the IgH 39 regulatory region. J. Exp. Med. 2014, 211, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Ghazzaui, N.; Issaoui, H.; Ferrad, M.; Carrion, C.; Cook-Moreau, J.; Denizot, Y.; Boyer, F. Eμ and 3’RR transcriptional enhancers of the IgH locus cooperate to promote c-myc-induced mature B-cell lymphomas. Blood Adv. 2020, 14, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Sabo, A.; Kress, T.R.; Pelizzola, M.; De Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Pisano, A.; Ceglia, S.; Palmieri, C.; Vecchio, E.; Fiume, G.; De Laurentiis, A.; Mimmi, S.; Falcone, C.; Iaccino, E.; Scialdone, A.; et al. CRL3IBTK regulates the tumor suppressor Pdcd4 through ubiquitylation coupled to proteasomal degradation. J. Biol. Chem. 2015, 290, 13958–13971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiume, G.; Scialdone, A.; Rizzo, F.; De Filippo, M.R.; Laudanna, C.; Albano, F.; Golino, G.; Vecchio, E.; Pontoriero, M.; Mimmi, S.; et al. IBTK Differently Modulates Gene Expression and RNA Splicing in HeLa and K562 Cells. Int. J. Mol. Sci. 2016, 17, 1848. [Google Scholar] [CrossRef] [Green Version]
- Albano, F.; Chiurazzi, F.; Mimmi, S.; Vecchio, E.; Pastore, A.; Cimmino, C.; Frieri, C.; Iaccino, E.; Pisano, A.; Golino, G.; et al. The expression of inhibitor of bruton’s tyrosine kinase gene is progressively up regulated in the clinical course of chronic lymphocytic leukaemia conferring resistance to apoptosis. Cell Death Dis. 2018, 1, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchio, E.; Golino, G.; Pisano, A.; Albano, F.; Falcone, C.; Ceglia, S.; Iaccino, E.; Mimmi, S.; Fiume, G.; Giurato, G.; et al. IBTK contributes to B-cell lymphomagenesis in Eμ-myc transgenic mice conferring resistance to apoptosis. Cell Death Dis. 2019, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Jiang, L.; Tseng, K.F.; Liu, Y.; Zhang, X.; Dong, R.; Zhigangc, L.; Xiujua, W. Aberrant NEAT1_1 expression may be a predictive marker of poor prognosis in diffuse large B cell lymphoma. Cancer Biomark. 2018, 23, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Liu, S.; Lu, S.; Yu, X.; Lai, J.; Wu, Y.; Chen, S.; Wang, L.; Yu, Z.; Luo, G.; et al. The c-Myc-regulated lncRNA NEAT1 and paraspeckles modulate imatinib-induced apoptosis in CML cells. Mol. Cancer 2018, 17, 130. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.S.; Li, L.J.; Huang, H.W.; Yang, H.F.; Wu, D.P. MYC-regulated lncRNA NEAT1 promotes B cell proliferation and lymphomagenesis via the miR-34b-5p-GLI1 pathway in diffuse large B-cell lymphoma. Cancer Cell. Int. 2020, 19, 20–87. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef]
- Craig, V.J.; Cogliatti, S.B.; Imig, J.; Renner, C.; Neuenschwander, S.; Rehrauer, H.; Schlapbach, R.; Dirnhofer, S.; Tzankov, A.; Müller, A. Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood 2011, 117, 6227–6236. [Google Scholar] [CrossRef]
- Christoffersen, N.R.; Shalgi, R.; Frankel, L.B.; Leucci, E.; Lees, M.; Klausen, M.; Pilpel, Y.; Nielsen, F.C.; Oren, M.; Lund, A.H. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010, 17, 236–245. [Google Scholar] [CrossRef]
- Aqeilan, R.I.; Calin, G.A.; Croce, C.M. miR-15a and miR-16-1 in cancer: Discovery, function and future perspectives. Cell Death Differ. 2010, 17, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsley, P.S.; Schelter, J.; Burchard, J.; Kibukawa, M.; Martin, M.M.; Bartz, S.R.; Johnson, J.M.; Cummins, J.M.; Raymond, C.K.; Dai, H.; et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol. Cell Biol. 2007, 27, 2240–2252. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, X.; Lin, J.; Lwin, T.; Wright, G.; Moscinski, L.C.; Dalton, W.S.; Seto, E.; Wright, K.; Sotomayor, E.; et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene 2012, 31, 3002–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Lwin, T.; Zhang, X.; Huang, A.; Wang, J.; Marquez, V.E.; Chen-Kiang, S.; Dalton, W.S.; Sotomayor, E.; Tao, J. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia 2013, 27, 2341–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell 2012, 22, 506–523. [Google Scholar] [CrossRef] [Green Version]
- Lenz, G.; Wright, G.W.; Emre, N.C.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; La, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520–13525. [Google Scholar] [CrossRef] [Green Version]
- Dal Bo, M.; Bomben, R.; Hernández, L.; Gattei, V. The MYC/miR-17-92 axis in lymphoproliferative disorders: A common pathway with therapeutic potential. Oncotarget. Review 2015, 14, 19381–19392. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Fassan, M.; Volinia, S.; Lovat, F.; Balatti, V.; Pekarsky, Y.; Croce, C.M. B-cell malignancies in microRNA Emu-miR-17~92 transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 18208–18213. [Google Scholar] [CrossRef] [Green Version]
- Oduor, C.I.; Kaymaz, Y.; Chelimo, K.; Otieno, J.A.; Ong’echa, J.M.; Moormann, A.M.; Bailey, J.A. Integrative microRNA and mRNA deep-sequencing expression profiling in endemic Burkitt lymphoma. BMC Cancer 2017, 17, 761. [Google Scholar] [CrossRef]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef]
- Boxer, L.M.; Dang, C.V. Translocations involving c-myc and c-myc function. Oncogene 2001, 20, 5595–5610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, V.; Bennett, M.J.; Walker, J.C.; Ma, C.; Jiang, I.; Cordon-Cardo, C.; Li, Q.J.; Lowe, S.W.; Hannon, G.J.; He, L. miR-19 is a key oncogenic component of mir-17-92. Genes Dev. 2009, 23, 2839–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, M.; Loosveld, M.; Montpellier, B.; Navarro, J.M.; Quilichini, B.; Picard, C.; DiCristofaro, J.; Bagnis, C.; Fossat, C.; Hernandez, L.; et al. Posttranscriptional deregulation of MYC via PTEN constitutes a major alternative pathway of MYC activation in T-cell acute lymphoblastic leukemia. Blood 2011, 117, 6650–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Zhang, Y.; Han, L.; Fu, L.; Mei, X.; Wang, J.; Itkow, J.; Elabid, A.E.I.; Pang, L.; Yu, D. Activating and sustaining c-Myc by depletion of miR-144/451 gene locus contributes to B-lymphomagenesis. Oncogene 2018, 37, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Pontoriero, M.; Fiume, G.; Vecchio, E.; De Laurentiis, A.; Albano, F.; Iaccino, E.; Mimmi, S.; Pisano, A.; Agosti, V.; Giovannone, E.; et al. Activation of NF-kB in B cell receptor signaling through Bruton’s tyrosine kinase-dependent phosphorylation of IκB-α. J. Mol. Med. 2019, 97, 675–690. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Interactor | Lymphoma Subtypes and Mouse Models | Targets of Interaction | Impact on Lymphomagenesis | References |
|---|---|---|---|---|
| lncRNA NEAT1 | DLBCL, CML | miR-34b-5p-GLI1 pathway | Reduction | [84,85] |
| PRDM11 | DLBCL | FOS and JUN | Reduction | [68] |
| MNT | vavP-MYC10 and Eμ-myc mice | BIM | Acceleration | [2,66] |
| FOXO3/FOXO1 | vavP-MYC10 and Eμ-myc mice BL | Reduction | [69,70] | |
| MAX | Eμ-myc mice | Reduction | [64] | |
| IBTK | Eμ-myc mice | MCL-1 and p53 | Acceleration | [67] |
| Eμ and 3′RR | c-myc-KIEμ, c-myc-KICμ, and c-myc-KICα mice BL-like lymphomas | Cooperate to induce | [78] |
| miRs | Functions | MYC Interactors | MYC Regulation | Lymphoma Subtypes | References |
|---|---|---|---|---|---|
| miR-34a | Tumor suppressor by promoting p53-dependent apoptosis | FOXP1 | Negative regulation | DLBCL,FL,GC-DLBCL | [87,88,90] |
| miR-15a/16-1 cluster | Tumor suppressor by targeting BCL2, Mcl-1, Cyclin D1 (miR-15a and miR 16-1); Tumor suppressor by inhibiting cell cycle-positive regulators including CDK6, Cyclins D and E (miR-16) | HDAC3 | Negative regulation | MCL | [91,92,93] |
| miR-26a | Tumorsuppressor by promoting apoptosis | EZH2 | Negative regulation | BL | [94] |
| miR-29 | Tumor suppressorby targeting CDK6, Mcl-1, IGF-1R | HDAC3, EZH2 | Negative regulation | MCL, GC-DLBCL, DLBCL, BL | [95] |
| miR-17-92 cluster | OncomiR by repressing the expression of BIM OncomiR by repressing the expression of PTEN (miR-19); OncomiR by down-regulating E2F1 (miR 17-20a) | Positive regulation | DLBCL, MCL, BL, GC-DLBCL | [96,97,98,99,100,101,102,103] | |
| miR144/451 | Positive feedback loop to safeguard the high level of MYC | Negative regulation | AML | [104] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vecchio, E.; Fiume, G.; Correnti, S.; Romano, S.; Iaccino, E.; Mimmi, S.; Maisano, D.; Nisticò, N.; Quinto, I. Insights about MYC and Apoptosis in B-Lymphomagenesis: An Update from Murine Models. Int. J. Mol. Sci. 2020, 21, 4265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124265
Vecchio E, Fiume G, Correnti S, Romano S, Iaccino E, Mimmi S, Maisano D, Nisticò N, Quinto I. Insights about MYC and Apoptosis in B-Lymphomagenesis: An Update from Murine Models. International Journal of Molecular Sciences. 2020; 21(12):4265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124265
Chicago/Turabian StyleVecchio, Eleonora, Giuseppe Fiume, Serena Correnti, Salvatore Romano, Enrico Iaccino, Selena Mimmi, Domenico Maisano, Nancy Nisticò, and Ileana Quinto. 2020. "Insights about MYC and Apoptosis in B-Lymphomagenesis: An Update from Murine Models" International Journal of Molecular Sciences 21, no. 12: 4265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124265