A Guide through the Dental Dimethacrylate Polymer Network Structural Characterization and Interpretation of Physico-Mechanical Properties


Abstract
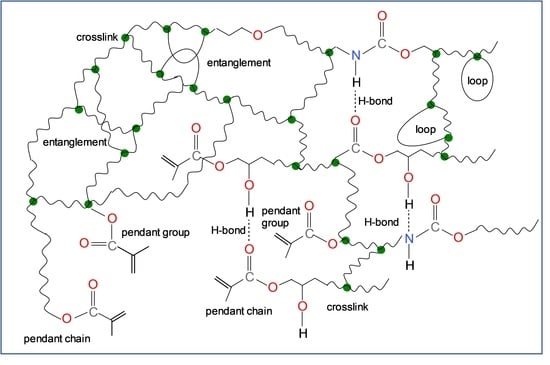
:
1. Introduction
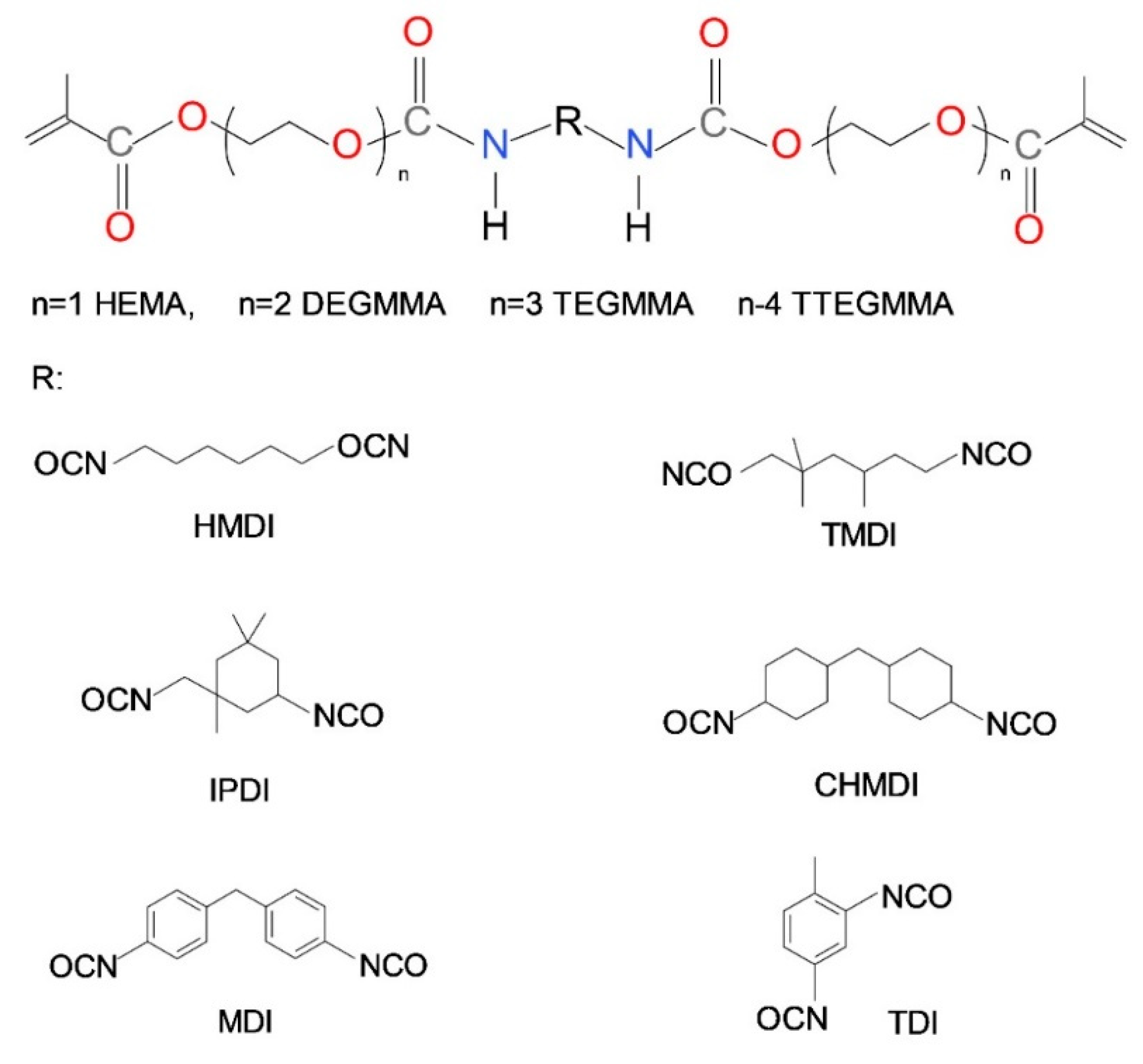
2. The Chemical Structure
3. The Chemical Crosslink Density
4. The Physical Crosslink Density
5. The Degree of Conversion
5.1. Methods of the DC Determination
5.1.1. Fourier Transformed Infrared (FTIR) Spectroscopy
5.1.2. Raman Spectroscopy (RS)
5.1.3. Differential Scanning Calorimetry (DSC)
5.1.4. Solid State Nuclear Magnetic Resonance (ssNMR)
5.2. The Influence of Chemical Structure on the DC
6. The Influence of Chemical Structure and Crosslink Density on Mechanical Properties
7. The Morphology
7.1. Qualitative Microscopic Characterization
7.2. Quantitative AFM Characterization
7.2.1. Phase Imagining
7.2.2. Roughness Analysis
7.2.3. Fractal Analysis
7.2.4. Percolation Theory
7.3. Quantitative XRPD Characterization
7.4. Thermal Analysis
8. Biocompatibility of Dental Dimethacrylate Polymer Networks
9. Conclusions
Funding
Conflicts of Interest
References
- Lovell, L.G.; Berchtold, K.A.; Elliot, J.E.; Lu, H.; Bowman, Ch.N. Understanding the kinetics and network formation of dimethacrylate dental resins. Polym. Adv. Technol. 2001, 12, 335–345. [Google Scholar] [CrossRef]
- Andrzejewska, E. Photopolymerization kinetics of multifunctional monomers. Prog. Polym. Sci. 2001, 26, 605–665. [Google Scholar] [CrossRef]
- Dickens, H.; Stansbury, J.W.; Choi, K.M.; Floyd, C.J.E. Photopolymerization kinetics of methacrylate dental resins. Macromolecules 2003, 36, 6043–6053. [Google Scholar] [CrossRef]
- Anseth, K.S.; Bowman, C.N. Kinetic gelation predictions of aggregation in tetrafunctional monomer polymerizations. J. Polym. Sci. B Polym. Phys. 1995, 33, 1769–1780. [Google Scholar] [CrossRef]
- Pfeifer, C.S.; Shelton, Z.R.; Braga, R.R.; Windmoller, D.; Machado, J.C.; Stansbury, J.W. Characterization of dimethacrylate polymeric networks: A study of the crosslinked structure formed by monomers used in dental composites. Eur. Polym. J. 2011, 47, 162–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, D.C. Dental Restorative Materials. In Materials Science and Technology: A Comprehensive Treatment; Cahn, R.W., Haasen, P., Kramer, E.J., Eds.; VCH: New York, NY, USA, 1992; Volume 14, pp. 209–258. [Google Scholar]
- Powers, J.M.; Sakaguchi, R.L. Restorative Materials—Composites and Polymers. In Craig’s Restorative Dental Materials, 13th ed.; Mosby: St. Louis, MI, USA, 2013; ISBN 9780323081085. [Google Scholar]
- Vasudeva, G. Monomer systems for dental composites and their future. J. Calif. Dent. Assoc. 2009, 37, 389–398. [Google Scholar] [PubMed]
- Anseth, K.; Newman, S.M.; Bowman, C.N. Polymeric Dental Composites: Properties and Reaction Behavior of Multimethacrylate Dental Restorations. In Biopolymers II. Advances in Polymer Science, 1st ed.; Peppas, N.A., Langer, R.S., Eds.; Springer: Berlin/Heidelberg, Germany, 1995; Volume 122, pp. 177–217. [Google Scholar]
- Astudillo-Rubio, D.; Delgado-Gaete, A.; Bellot-Arcís, C.; Montiel-Company, J.M.; Pascual-Moscardó, A.; Almerich-Silla, J.M. Mechanical properties of provisional dental materials: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0193162. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.H.S.; Mai, Y.; Kim, H.; Tong, K.C.T.; Ng, D.; Hsiao, J.C.M. Review: Resin composite filling. Materials 2010, 3, 1228–1243. [Google Scholar] [CrossRef]
- Kwon, T.Y.; Bagheri, R.; Kim, Y.K.; Kim, K.H.; Burrow, M.F. Cure mechanisms in materials for use in esthetic dentistry. J. Investig. Clin. Dent. 2012, 3, 3–16. [Google Scholar] [CrossRef]
- Singh, A.V.; Ansari, M.H.D.; Laux, P.; Luch, A. Micro-nanorobots: Important considerations when developing novel drug delivery platforms. Expert Opin. Drug Deliv. 2019, 16, 1259–1275. [Google Scholar] [CrossRef]
- Santulli, C. Nanostructured Composites for Dental Fillings. In Nanostructured Polymer Composites for Biomedical Applications; Swain, S.K., Jawaid, M., Eds.; Elsevier: Amsterdam, NL, USA; Oxford, UK; Cambridge, UK, 2019; pp. 277–294. [Google Scholar]
- Bowen, R.L. Dental Filling Material Comprising Vinyl Silane Treated Fused Silica and a Binder Consisting of the Reaction Product of Bis Phenol and Glycidyl Acrylate. U.S. Patent 3066112A, 26 June 1962. [Google Scholar]
- Peutzfeldt, A. Resin composites in dentistry: The monomer systems. Eur. J. Oral. Sci. 1997, 105, 97–116. [Google Scholar] [CrossRef] [PubMed]
- El-Banna, A.; Sherief, D.; Fawzy, A.S. Resin Based Dental Composites for Tooth Filling. In Advanced Dental Biomaterials; Khurshid, Z., Najeeb, S., Zafar, M.S., Sefat, F., Eds.; Elsevier: Duxford, UK; Cambridge, UK; Kidlington, UK, 2019; pp. 127–174. [Google Scholar]
- Sideridou, I.; Tserki, V.; Papanastasiou, G. Effect of chemical structure on degree of conversion in light-cured dimethacrylate-based dental resins. Biomaterials 2002, 23, 1819–1829. [Google Scholar] [CrossRef]
- Gajewski, V.E.S.; Pfeifer, C.S.; Fróes-Salgado, N.R.G.; Boaro, L.C.C.; Braga, R.R. Monomers used in resin composites: Degree of conversion, mechanical properties and water sorption/solubility. Braz. Dent. J. 2012, 23, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Dusek, K.; MacKnight, W. Cross-Linking and Structure of Polymer Networks. In Crosslinked Polymers: Chemistry, Properties, and Applications; Dickie, R.A., Labana, S.S., Bauer, R.S., Eds.; American Chemical Society: Washington, DC, USA, 1988; Volume 1, pp. 2–27. [Google Scholar]
- Hild, G. Model networks based on ‘endlinking’ processes: Synthesis, structure and properties. Prog. Polym. Sci. 1998, 23, 1019–1149. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Theory of Rubber Elasticity. Polymer 1985, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.E.; Lovell, L.G.; Bowman, C.N. Primary cyclization in the polymerization of bis-GMA and TEGDMA: A modeling approach to understanding the cure of dental resins. Dent. Mater. 2001, 17, 221–229. [Google Scholar] [CrossRef]
- Roshchupkin, V.; Kumaz, S. Network Structure Formation (Radical polymerization). In Polymeric Materials Encyclopedia, 1st ed.; Salamone, J.C., Ed.; CRC Press: Boca Raton, FL, USA, 1996; Volume 6, pp. 4576–4581. [Google Scholar]
- Kannurpatti, A.; Anseth, J.; Bowman, C.H.N. A study of the evolution of mechanical properties and structural heterogeneity of polymer networks formed by photo-polymerizations of multifunctional (meth)acrylates. Polymer 1998, 39, 2507–2513. [Google Scholar] [CrossRef]
- Rey, L.; Duchet, J.; Galy, J.; Sautereau, H.; Vouagner, D.; Carrion, L. Structural heterogeneities and mechanical properties of vinyl/dimethacrylate networks synthesized by thermal free radical polymerization. Polymer 2002, 43, 4375–4384. [Google Scholar] [CrossRef]
- Cook, W.D. Fracture and structure of highly crosslinked polymer composites. J. Appl. Polym. Sci. 1991, 42, 1259–1269. [Google Scholar] [CrossRef]
- Simon, I.G.P.; Allen, P.E.M.; Williams, D.R.G. Properties of dimethacrylate copolymers of varying crosslink density. Polymer 1991, 32, 2577–2587. [Google Scholar] [CrossRef]
- Wang, E.; Hasheminasab, A.; Guo, Y.; Soucek, M.D.; Cakmak, M. Structure characterization of UV-curing PEG-b-PPG-b-PEG dimethacrylate cross-linked network. Polymer 2018, 153, 241–249. [Google Scholar] [CrossRef]
- Gu, Y.; Zhao, J.; Johnson, J.A. A (Macro)Molecular-level understanding of polymer network topology. Trends Chem. 2019, 1, 318–334. [Google Scholar] [CrossRef]
- Stansbury, J.W. Dimethacrylate network formation and polymer property evolution as determined by the selection of monomers and curing conditions. Dent. Mater. 2012, 28, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barszczewska-Rybarek, I. Structure-property relationships in dimethacrylate networks based on Bis-GMA, UDMA and TEGDMA. Dent. Mater. 2009, 25, 1082–1089. [Google Scholar] [CrossRef]
- Barszczewska-Rybarek, I.; Krasowska, M. Fractal analysis of heterogeneous polymer networks formed by photopolymerization of dental dimethacrylates. Dent. Mater. 2012, 28, 695–702. [Google Scholar] [CrossRef]
- Krasowska, M.; Barszczewska-Rybarek, I. The percolation theory in studying the morphology of polymer networks formed by photopolymerization of dental dimethacrylates. Eur. Polym. J. 2016, 76, 77–78. [Google Scholar] [CrossRef]
- Barszczewska-Rybarek, I. The role of molecular structure on impact resistance and bending strength of photocured urethane-dimethacrylate polymer networks. Polym. Bull. 2017, 74, 4023–4040. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Seiffert, S. Nanostructural heterogeneity in polymer networks and gels. Polym Chem 2015, 6, 5515–5528. [Google Scholar] [CrossRef]
- Krzeminski, M.; Molinari, M.; Defoort, B.; Coqueret, X. Nanoscale heterogeneities in radiation-cured diacrylate networks: Weakness or asset? Radiat. Phys. Chem. 2013, 84, 79–84. [Google Scholar] [CrossRef]
- Krzeminski, M.; Molinari, M.; Troyon, M.; Coqueret, X. Characterization by atomic force microscopy of the nanoheterogeneities produced by the radiation-induced cross-linking polymerization of aromatic diacrylates. Macromolecules 2010, 43, 8121–8127. [Google Scholar] [CrossRef]
- Krzeminski, M.; Molinari, M.; Troyon, M.; Coqueret, X. Calorimetric characterization of the heterogeneities produced by the radiation-induced cross-linking polymerization of aromatic diacrylates. Macromolecules 2010, 43, 3757–3763. [Google Scholar] [CrossRef]
- Panyukov, S.V. Theory of heterogeneities in polymer networks. Polym. Sci. A 2016, 58, 886–898. [Google Scholar] [CrossRef]
- Seiffer, S. Origin of nanostructural inhomogeneity in polymer-network gels. Polym. Chem. 2017, 8, 4472–4487. [Google Scholar] [CrossRef]
- Guo, Z.; Sautereau, H.; Kranbuehl, D. Evidence for spatial heterogeneities observed by frequency dependent dielectric and mechanical measurements in vinyl/dimethacrylate systems. Polymer 2010, 26, 416–425. [Google Scholar] [CrossRef]
- Husar, B.; Commereuc, S.; Chmela, S.; Verney, V. Characterization of networks from photoreactive copolymers: An attempt to correlate chemical composition to network structure. Polym. Int. 2010, 59, 1563–1570. [Google Scholar] [CrossRef]
- Szczepanski, R.C.; Pfeifer, C.S.; Stansbury, J.W. A new approach to network heterogeneity: Polymerization induced phase separation in photo-initiated, free-radical methacrylic systems. Polymer 2012, 53, 4694–4701. [Google Scholar] [CrossRef] [Green Version]
- Barszczewska-Rybarek, I.M. A new approach to morphology studies on diacrylate polymer networks using X-ray powder diffraction. Macromol. Chem. Phys. 2013, 214, 1019–1026. [Google Scholar] [CrossRef]
- Moore, R.J.; Watts, J.T.F.; Hood, J.A.A.; Burritt, D.J. Intra-oral temperature variation over 24 hours. Eur. J. Orthod. 1999, 21, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Leprince, J.G.; Palin, W.M.; Hadis, M.A.; Devaux, J.; Leloup, G. Progress in dimethacrylate-based dental composite technology and curing efficiency. Dent. Mater. 2013, 29, 139–156. [Google Scholar] [CrossRef]
- Barszczewska-Rybarek, I.M. Characterization of urethane-dimethacrylate derivatives as alternative monomers for the restorative composite matrix. Dent. Mater. 2014, 30, 1336–1344. [Google Scholar] [CrossRef]
- Lee, D.W.; Kim, H.N.; Lee, D.S. Introduction of reversible urethane bonds based on vanillyl alcohol for efficient self-healing of polyurethane elastomers. Molecules 2019, 24, 2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemon, M.T.; Jones, M.S.; Stansbury, J.W. Hydrogen bonding interactions in methacrylate monomers and polymers. J. Biomed. Mater. Res. A 2007, 83, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Kanlayakan, N.; Kerdpol, K.; Prommin, C.; Salaeh, R.; Chansen, W.; Sattayanon, C.; Kungwan, N. Effects of different proton donor and acceptor groups on excited-state intramolecular proton transfers of amino-type and hydroxy-type hydrogen-bonding molecules: Theoretical insights. New J. Chem 2017, 41, 8761–8771. [Google Scholar] [CrossRef]
- Park, J.; Eslick, J.; Ye, Q.; Misra, A.; Spencer, P. The influence of chemical structure on the properties in methacrylate-based dentin adhesives. Dent. Mater. 2011, 27, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Sideridou, I.; Tserki, V.; Papanastasiou, G. Study of water sorption, solubility and modulus of elasticity of light-cured dimethacrylate-based dental resins. Biomaterials 2003, 24, 655–665. [Google Scholar] [CrossRef]
- Ogliari, F.A.; Ely, C.; Zanchi, C.H.; Fortes, C.B.; Samuel, S.M.; Demarco, F.F.; Petzhold, C.L.; Piva, E. Influence of chain extender length of aromatic dimethacrylates on polymer network development. Dent. Mater. 2007, 24, 165–171. [Google Scholar] [CrossRef]
- Barszczewska-Rybarek, I.; Jurczyk, S. Comparative Study of Structure-Property Relationships in Polymer Networks Based on Bis-GMA, TEGDMA and Various Urethane-Dimethacrylates. Materials 2015, 8, 1230–1248. [Google Scholar] [CrossRef] [Green Version]
- Asmussen, E.; Peutzfeldt, A. Influence of UEDMA, BisGMA, and TEGDMA on selected mechanical properties of experimental resin composites. Dent. Mater. 1998, 14, 51–56. [Google Scholar] [CrossRef]
- Bociong, K.; Szczesio, A.; Sokolowski, K.; Domarecka, M.; Sokolowski, J.; Krasowski, M.; Lukomska-Szymanska, M. The influence of water sorption of dental light-cured composites on shrinkage stress. Materials 2017, 10, 1142. [Google Scholar] [CrossRef]
- Sideridou, I.; Achilias, D.S.; Spyroudi, C.; Karabela, M. Water sorption characteristics of light-cured dental resins and composites based on Bis-EMA/PCDMA. Biomaterials 2004, 25, 367–376. [Google Scholar] [CrossRef]
- Łukaszczyk, J.; Janicki, B.; Frick, A. Investigation on synthesis and properties of isosorbide based bis-GMA analogue. J. Mater. Sci. Mater. Med. 2012, 23, 1149–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, L. Proton donor is more important than proton acceptor in hydrogen bond formation: A universal equation for calculation of hydrogen bond strength. Phys. Chem. A 2003, 107, 11517–11524. [Google Scholar] [CrossRef]
- Jeffrey, G. An Introduction to Hydrogen Bonding, 1st ed.; Oxford University Press: New York, NY, USA, 1997; pp. 33–55. [Google Scholar]
- Barszczewska-Rybarek, I.M.; Korytkowska-Wałach, A.; Kurcok, M.; Chladek, G.; Kasperski, J. DMA analysis of the structure of crosslinked poly(methyl methacrylate)s. Acta. Bioeng. Biomech. 2017, 19, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Reiche, A.; Sandner, R.; Weinkauf, A.; Sandner, B.; Fleischer, G.; Rittig, F. Gel electrolytes on the basis of oligo(ethylene glycol) (n) dimethacrylates—Thermal, mechanical and electrochemical properties in relationship to the network structure. Polymer 2000, 41, 3821–3836. [Google Scholar] [CrossRef]
- Barszczewska-Rybarek, I. Prediction of physical properties of dimethacrylate polymer networks by a group contribution approach. Int. J. Polym. Anal. Charact 2013, 18, 93–104. [Google Scholar] [CrossRef]
- Barszczewska-Rybarek, I.; Korytkowska, A.; Gibas, M. Investigations on the structure of poly(dimethacrylate)s. Des. Monomers. Polym. 2001, 4, 301–314. [Google Scholar] [CrossRef]
- Khatri, C.A.; Stansbury, J.W.; Schultheisz, C.R.; Antonucci, J.M. Synthesis, characterization and evaluation of urethane derivatives of Bis-GMA. Dent. Mater. 2003, 19, 584–588. [Google Scholar] [CrossRef]
- Ge, J.; Trujillo, M.; Stansbury, J.W. Synthesis and photopolymerization of low shrinkage methacrylate monomers containing bulky substituent groups. Dent. Mater. 2005, 21, 1163–1169. [Google Scholar] [CrossRef]
- Barszczewska-Rybarek, I.; Gibas, M.; Kurcok, M. Evaluation of the network parameter in aliphatic poly(urethane dimethacrylate)s by dynamic thermal analysis. Polymer 2000, 41, 3129–3135. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Dias, A.A. Analysis of network structure of UV-cured acrylates by 1H NMR relaxation, 13C NMR spectroscopy, and dynamic mechanical experiments. Macromolecules 2001, 34, 4051–4060. [Google Scholar] [CrossRef]
- Borges, M.G.; Barcelos, L.M.; Menezes, M.S.; Soares, C.J.; Fugolin, A.P.P.; Navarro, O.; Huynh, V.; Lewis, S.H.; Pfeifer, C.S. Effect of the addition of thiourethane oligomers on the sol–gel composition of BisGMA/TEGDMA polymer networks. Dent. Mater. 2019, 35, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Béhin, P.; Stoclet, G.; Ruse, D.; Sadoun, M. Dynamic mechanical analysis of high pressure polymerized urethane dimethacrylate. Dent. Mater. 2014, 30, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yu, P.; Dong, S. The influence of crosslink density on the failure behavior in amorphous polymers by molecular dynamics simulations. Materials 2016, 9, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malana, M.A.; Bukhari, J.D.; Zohra, R. Synthesis, swelling behavior, and network parameters of novel chemically crosslinked poly (acrylamide-co-methacrylate-co-acrylic acid) hydrogels. Des. Monom. Polym 2014, 17, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Flory, P.J.; Rehner, J. Statistical mechanics of crosslinked polymers. Rubber like elasticity. J. Chem. Phys. 1943, 11, 521. [Google Scholar] [CrossRef]
- Alger, M.S.M. Polymer Science Dictionary, 3rd ed.; Springer: Berlin, Germany, 2017. [Google Scholar]
- Redman, R.P. Developments in Polyurethane Elastomers. In Developments in Polyurethane; Buist, J.M., Ed.; Applied Science Publishers: London, UK, 1978; pp. 33–176. [Google Scholar]
- Gonçalves, F.; Kawano, Y.; Pfeifer, C.S.; Stansbury, J.W.; Braga, R.R. Influence of bis-GMA, TEGDMA, and bis-EMA contents on viscosity, conversion, and flexural strength of experimental resins and composites. Eur. J. Oral. Sci. 2009, 117, 442–446. [Google Scholar] [CrossRef]
- Baroudi, K.; Saleh, A.M.; Silikas, N.; Watts, D.C. Shrinkage behaviour of flowable resin-composites related to conversion and filler-fraction. J. Dent. 2007, 35, 651–655. [Google Scholar] [CrossRef]
- Benesi, H.A.; Hildebrand, J.H. A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons. J. Amer. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Yilgör, E.; Burgaz, E.; Yurtsever, E.; Yilgör, İ. Comparison of hydrogen bonding in polydimethylsiloxane and polyether based urethane and urea copolymers. Polymer 2000, 41, 849–857. [Google Scholar] [CrossRef]
- Antonucci, J.M.; Fowler, B.O.; Weir, M.D.; Skrtic, D.; Stansbury, J.W. Effect of ethyl-alpha-hydroxymethylacrylate on selected properties of copolymers and ACP resin composites. J. Mater. Sci. Mater. Med. 2008, 19, 3263–3271. [Google Scholar] [CrossRef] [Green Version]
- Alshali, R.Z.; Silikas, N.; Satterthwaite, J.D. Degree of conversion of bulk-fill compared to conventional resin-composites at two time intervals. Dent. Mater. 2013, 29, e213–e217. [Google Scholar] [CrossRef] [PubMed]
- Randolph, L.D.; Palin, W.M.; Bebelman, S.; Devaux, J.; Gallez, B.; Leloup, G.; Leprince, J.G. Ultra-fast light-curing resin composite with increased conversion and reduced monomer elution. Dent. Mater. 2014, 30, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Par, M.; Gamulin, O.; Marovic, D.; Klaric, E.; Tarle, Z. Raman spectroscopic assessment of degree of conversion of bulk-fill resin composites—Changes at 24 hours post cure. Oper. Dent. 2015, 40, E92–E101. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, M.; Balazsi, R.; Soanca, A.; Roman, A.; Sarosi, C.; Prodan, D.; Vlassa, M.; Cojocaru, I.; Saceleanu, V.; Cristescu, I. Evaluation of the degree of conversion, residual monomers and mechanical properties of some light-cured dental resin composites. Materials 2019, 12, 2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackert, J.R. Physical Properties and Biocompatibility. In Dental Materials and Their Selection, 4th ed.; O’Brien, W.J., Ed.; Quintessence Publishing: Chicago, IL, USA, 2009; pp. 12–24. [Google Scholar]
- Roman, A.; Páll, E.; Moldovan, M.; Rusu, D.; Soritau, O.; Festila, D.; Lupse, M. Cytotoxicity of experimental resin composites on Mesenchymal Stem Cells isolated from two oral sources. Microsc. Microanal. 2016, 22, 1018–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manhart, J.; Chen, H.; Hamm, G.; Hickel, R. Buonocore Memorial Lecture. Review of the clinical survival of direct and indirect restorations in posterior teeth of the permanent dentition. Oper. Dent. 2004, 29, 481–508. [Google Scholar]
- Lempel, E.; Czibulya, Z.; Kovács, B.; Szalma, J.; Tóth, Á.; Kunsági-Máté, S.; Varga, Z.; Böddi, K. Degree of Conversion and BisGMA, TEGDMA, UDMA Elution from Flowable Bulk Fill Composites. Int. J. Mol. Sci. 2016, 17, 732. [Google Scholar] [CrossRef] [Green Version]
- Barszczewska-Rybarek, I.M. Quantitative determination of degree of conversion in photocured poly(urethane-dimethacrylate)s by FTIR spectroscopy. J. Appl. Polym. Sci. 2012, 123, 1604–1611. [Google Scholar] [CrossRef]
- Collares, F.M.; Portella, F.F.; Leitune, V.C.B.; Samuel, S.M.W. Discrepancies in degree of conversion measurements by FTIR. Braz. Oral. Res. 2014, 28, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Barszczewska-Rybarek, I.; Chladek, G. Studies on the curing efficiency and mechanical properties of Bis-GMA and TEGDMA nanocomposites containing silver nanoparticles. Int. J. Mol. Sci. 2018, 19, 3937. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kranbuehl, D.E.; Sautereau, H.; Seytre, G.; Dupuy, J. Modelling and measuring UV cure kinetics of thick dimethacrylate samples. Int. J. Biol. Macromol. 2009, 42, 203–210. [Google Scholar] [CrossRef]
- Guimaraes, T.; Schneider, L.F.; Braga, R.R.; Pfeifer, C.S. Mapping camphorquinone consumption, conversion and mechanical properties in methacrylates with systematically varied CQ/amine compositions. Dent. Mater. 2014, 30, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, W.F.; Vallo, C. Effect of different photoinitiator systems on conversion profiles of a model unfilled light-cured resin. Dent. Mater. 2007, 23, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Emami, N.; Söderholm, K.J. Influence of light-curing procedures and photoinitiator/co-initiator composition on the degree of conversion of light-curing resins. J. Mater. Sci. Mater. Med. 2005, 16, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Denis, A.B.; Diagone, C.A.; Plepis, A.M.G.; Viana, R.B. The effect of the polymerization initiator and light source on the elution of residual BisGMA and TEGDMA monomers: A study using liquid chromatography with UV detection. Spectr. Acta A Mol. Biomol. Spectr. 2015, 151, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Suh, B.I.; Shin, D.; Kim, K.M. Comparison of the Physical and Mechanical Properties of Resin Matrix with Two Photoinitiator Systems in Dental Adhesives. Polymers 2016, 8, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halvorson, R.H.; Erickson, R.L.; Davidson, C.L. The effect of filler and silane content on conversion of resin-based composite. Dent. Mater. 2003, 19, 327–333. [Google Scholar] [CrossRef]
- Sahebalam, R.; Boruziniat, A.; Mohammadzadeh, F.; Rangrazi, A. Effect of the time of salivary contamination during light curing on degree of conversion and microhardness of a restorative composite resin. Biomimetics 2018, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Moraes, L.G.; Rocha, R.S.; Menegazzo, L.M.; de Araújo, E.B.; Yukimito, K.; Moraes, J.C. Infrared spectroscopy: A tool for determination of the degree of conversion in dental composites. J. Appl. Oral. Sci. 2008, 16, 145–149. [Google Scholar] [CrossRef]
- Pfeifer, C.S.; Silva, L.R.; Kawano, Y.; Braga, R.R. Bis-GMA co-polymerizations: Influence on conversion, flexural properties, fracture toughness and susceptibility to ethanol degradation of experimental composites. Dent. Mater. 2009, 25, 1136–1141. [Google Scholar] [CrossRef]
- Stencel, R.; Pakieła, W.; Barszczewska-rybarek, I.; Żmudzki, J.; Kasperski, J.; Chladek, G. Effects of different inorganic fillers on mechanical properties and degree of conversion of dental resin composites. Arch. Metall. Mater. 2018, 63, 1361–1369. [Google Scholar] [CrossRef]
- Zhang, M.; Puska, M.A.; Botelho, M.G.; Säilynoja, E.S.; Matinlinna, J.P. Degree of conversion and leached monomers of urethane dimethacrylate-hydroxypropyl methacrylate-based dental resin systems. J. Oral. Sci. 2016, 58, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Liu, F.; He, J. Preparation and characterization of low polymerization shrinkage and Bis-GMA-free dental resin system. Adv. Polym. Technol. 2015, 34, 21503. [Google Scholar] [CrossRef]
- Scherzer, T.; Tauber, A.; Mehnert, R. UV curing of pressure sensitive adhesives studied by real-time FTIR-ATR spectroscopy. Vib. Spectrosc. 2002, 29, 125–131. [Google Scholar] [CrossRef]
- Al-Odayni, A.B.; Alfotawi, R.; Khan, R.; Saeed, W.S.; Al-Kahtani, A.; Aouak, T.; Alrahlah, A. Synthesis of chemically modified BisGMA analog with low viscosity and potential physical and biological properties for dental resin composite. Dent. Mater. 2019, 35, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Scherzer, T. Real-time FTIR-ATR spectroscopy of photopolymerization reactions. Macromol. Symp. 2002, 184, 79–98. [Google Scholar] [CrossRef]
- Fringeli, U.P. ATR and Reflectance IR Spectroscopy, Applications. In Encyclopedia of Spectroscopy and Spectrometry, 3rd ed.; Lindon, J.C., Tranter, G.E., Koppenaal, D.W., Eds.; Academic Press: Kidlington, UK, 2017; pp. 115–129. [Google Scholar] [CrossRef]
- Ferrer, N. Forensic Science, Applications of IR Spectroscopy, In Encyclopedia of Spectroscopy and Spectrometry, 3rd ed.; Lindon, J.C., Tranter, G.E., Koppenaal, D.W., Eds.; Academic Press: Kidlington, UK, 2017; pp. 695–706. [Google Scholar] [CrossRef]
- Griffiths, P. FTIR vs. FT-IR vs. Mid-IR. Appl. Spectrosc. 2010, 64, 40A. [Google Scholar] [CrossRef]
- Lin-Vien, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Academic Press: London, UK, 1991; pp. 137–281. [Google Scholar]
- Saen, P.; Atai, M.; Nodehi, A.; Solhi, L. Physical characterization of unfilled and nanofilled dental resins: Static versus dynamic mechanical properties. Dent. Mater. 2016, 32, e185–e197. [Google Scholar] [CrossRef]
- Yu, B.A.; Liu, D.L.; Liu, F.; He, J.W. Preparation and characterization of light-cured dental resins without methacrylate monomers derived from bisphenol A. Adv. Polym. Technol. 2016, 33, 21417. [Google Scholar] [CrossRef]
- da Silva, E.M.; Miragaya, L.; Noronha-Filho, J.D.; Amaral, C.M.; Poskus, L.T.; Guimarães, J.G.A. Characterization of an experimental resin composite organic matrix based on a tri-functional methacrylate monomer. Dent. Mater. J. 2016, 35, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Pereira, S.; Nunes, T.; Kalachandra, S. Low viscosity dimethacrylate comonomer compositions [Bis-GMA and CH3Bis-GMA] for novel dental composites; analysis of the network by stray-field MRI, solid-state NMR and DSC & FTIR. Biomaterials 2002, 23, 3799–3806. [Google Scholar] [CrossRef] [PubMed]
- Sideridou, I.D.; Karabela, M.M. Effect of the amount of 3-methacryloxypropyltrimethoxysilane coupling agent on physical properties of dental resin nanocomposites. Dent. Mater. 2009, 25, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Podgórski, M. Structure–property relationship in new photo-cured dimethacrylate based dental resins. Dent. Mater. 2012, 28, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Podgorski, M. Synthesis and characterization of novel dimethacrylates of different chain lengths as possible dental resins. Dent. Mater. 2010, 26, e188–e194. [Google Scholar] [CrossRef]
- Gauthier, M.A.; Stangel, I.; Ellis, T.H.; Zhu, X.X. A new method for quantifying the intensity of the C=C band of dimethacrylate dental monomers in their FTIR and Raman spectra. Biomaterials. 2005, 26, 6440–6448. [Google Scholar] [CrossRef]
- Stansbury, J.W.; Dickens, S.H. Determination of double bond conversion in dental resins by near infrared spectroscopy. Dent. Mater. 2001, 17, 71–79. [Google Scholar] [CrossRef]
- Floyd, C.J.; Dickens, S.H. Network structure of Bis-GMA- and UDMA-based resin systems. Dent. Mater. 2006, 22, 1143–1149. [Google Scholar] [CrossRef]
- Lungu, A.; Şulcă, N.M.; Vasile, E.; Badea, N.; Pârvu, C.; Iovu, H. The influence of POSS substituent on synthesis and properties of hybrid materials based on urethane dimethacrylate (UDMA) and various polyhedral oligomeric silsesquioxane (POSS). J. Appl. Polym. Sci. 2011, 121, 2919–2926. [Google Scholar] [CrossRef]
- Johnck, M.; Muller, L.; Neyer, A.; Hofstraat, J.W. Quantitative determination of unsaturation in photocured halogenated acrylates and methacrylates by FT-IR and Raman-spectroscopy and by thermal analysis. Polymer 1999, 40, 3631–3640. [Google Scholar] [CrossRef]
- Luo, S.; Zhu, W.; Liu, F.; He, J. Preparation of a Bis-GMA-free dental resin system with synthesized fluorinated dimethacrylate monomers. Int. J. Mol. Sci. 2016, 17, 2014. [Google Scholar] [CrossRef] [Green Version]
- Kammer, S.; Albinsky, K.; Sandner, B.; Wartewig, S. Polymerization of hydroxyalkyl methacrylates characterized by combination of FT-Raman and step-scan FT-i.r. photoacoustic spectroscopy. Polymer 1999, 40, 1131–1137. [Google Scholar] [CrossRef]
- Rueggeberg, FA. Determination of resin cure using infrared analysis without an internal standard. Dent. Mater. 1994, 10, 282–286. [Google Scholar] [CrossRef]
- Silva Soares, L.E.; Martin, A.A.; Barbosa Pinheiro, A.L. Degree of conversion of composite resin: A Raman study. J. Clin. Laser Med. Surg. 2003, 21, 357–362. [Google Scholar] [CrossRef] [PubMed]
- BinMahfooz, A.M.; Qutub, O.A.; Marghalani, T.Y.; Ayad, M.F.; Maghrabi, A.A. Degree of conversion of resin cement with varying methacrylate compositions used to cement fiber dowels: A Raman spectroscopy study. J. Prosthet. Dent. 2018, 119, 1014–1020. [Google Scholar] [CrossRef]
- Pianelli, C.; Devaux, J.; Bebelman, S.; Leloup, G. The micro-Raman spectroscopy, a useful tool to determine the degree of conversion of light-activated composite resins. J. Biomed. Mater. Res. 1999, 48, 675–681. [Google Scholar] [CrossRef]
- Habib, E.; Zhu, X.X. Photo-calorimetry method optimization for the study of light-initiated radical polymerization of dental resins. Polymer 2018, 135, 178–184. [Google Scholar] [CrossRef]
- Jakubiak, J.; Sionkowska, A.; Lindén, L.Å.; Rabek, F. Isothermal Photo differential scanning calorimetry. Crosslinking polymerization of multifunctional monomers in presence of visible light photoinitiators. J. Therm. Anal. Calorim. 2001, 65, 435. [Google Scholar] [CrossRef]
- Morancho, J.M.; Cadenato, A.; Fernández-Francos, X.; Salla, J.M.; Ramis, X. Isothermal kinetics of photopolymerization and thermal polymerization of Bis-gma/TEGDMA resins. J. Therm. Anal. Calorim. 2008, 98, 513–522. [Google Scholar] [CrossRef]
- Antonucci, J.M.; Toth, E.E. Extent of polymerization of dental resins by differential scanning calorimetry. J. Dent. Res. 1983, 62, 121–125. [Google Scholar] [CrossRef]
- Moszner, N.; Völkel, T.; Fischer, U.K.; Klester, A.; Rheinberger, V. Synthesis and polymerisation of new multifunctional urethane methacrylates. Angew Makromol. Chem. 1999, 265, 31–35. [Google Scholar] [CrossRef]
- Barszczewska-Rybarek, I. Study on the effects of urethane-dimethacrylates’ structures on the morphology and properties of polymers based on them. Polimery 2008, 53, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Sawada, H. Thermodynamics of Polymerization. I. J. Macromol. Sci. C 1969, 3, 313–338. [Google Scholar] [CrossRef]
- Morgan, D.R.; Kalachandra, S.; Shobha, H.K.; Gunduz, N.; Stejskal, E.O. Analysis of a dimethacrylate copolymer (BisGMA and TEGDMA) network by DSC and 13C solution and solid-state NMR spectroscopy. Biomaterials 2000, 21, 1897–1903. [Google Scholar] [CrossRef]
- Lopez-Suevos, F.; Dickens, S.H. Degree of cure and fracture properties of experimental acid-resin modified composites under wet and dry conditions. Dent. Mater. 2008, 24, 778–785. [Google Scholar] [CrossRef] [Green Version]
- Jancar, J.; Wang, W.; DiBenedetto, A.T. On the heterogeneous structure of thermally cured bis-GMA/TEGDMA resins. J. Mater. Sci. Mater. Med. 2000, 11, 675–682. [Google Scholar] [CrossRef]
- Wool, R.P. Properties of Triglyceride-Based Thermosets. In Bio-Based Polymers and Composites; Wool, R.P., Sun, X.S., Eds.; Elsevier Academic Press: Boston, MA, USA, 2005; pp. 202–255. [Google Scholar] [CrossRef]
- Ferracane, J. Correlation between hardness and degree of conversion during the setting reaction of unfilled dental restorative resins. Dent. Mater. 1985, 1, 11–14. [Google Scholar] [CrossRef]
- Asmussen, E. Restorative resins: Hardness and strength vs. quantity of remaining double bonds. Scand. J. Dent. Res. 1982, 90, 484–489. [Google Scholar] [CrossRef]
- Linden, L.A. Dental Polymers (Overview). In Polymeric Materials Encyclopedia, 1st ed.; Salamone, J.C., Ed.; CRC Press: Boca Raton, FL, USA, 1996; Volume 12, p. 1849. [Google Scholar]
- Badakar, C.M.; Shashibhushan, K.K.; Naik, N.S.; Reddy, V.V. Fracture resistance of microhybrid composite, nano composite and fibre-reinforced composite used for incisal edge restoration. Dent. Traumatol. 2011, 27, 225–229. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamazaki, M.; Kobiki, Y. Direct observation of polymer gel surfaces by atomic force microscopy. J. Chem. Phys. 1996, 104, 1751–1757. [Google Scholar] [CrossRef]
- Caglayan, M.O. Nanomechanical characterization of flowable dental restorative nanocomposite resins using AFM. Polym. Plast. Technol. Eng. 2017, 56, 1813–1821. [Google Scholar] [CrossRef]
- Munz, M. Microstructure and roughness of photopolymerized poly(ethylene glycol) diacrylate hydrogel as measured by atomic force microscopy in amplitude and frequency modulation mode. Appl. Surf. Sci. 2013, 279, 300–309. [Google Scholar] [CrossRef]
- Gurtovenko, A.A.; Gotlib, Y.; Kilian, H. Viscoelastic dynamic properties of heterogeneous polymer networks with domain structure. Macromol. Theory Simul. 2000, 9, 388–397. [Google Scholar] [CrossRef]
- Gotlib, Y.; Gurtovenko, A.A.; Kilian, H.G. Relaxation modulus of heterogeneous polymer networks with the domain structure. Polym. Sci. A 2001, 43, 308–314. [Google Scholar]
- Sideridou, I.D.; Karabela, M.M.; Vouvoudi, E.C. Dynamic thermo-mechanical properties and sorption characteristics of two commercial light cured dental resin composites. Dent. Mater. 2008, 24, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Dean, K.M.; Cook, W.D. Small angle neutron scattering and dynamic mechanical thermal analysis of dimethacrylate/epoxy IPNs. Eur. Polym. J. 2006, 42, 2872–2887. [Google Scholar] [CrossRef]
- Abedin, F.; Roughton, B.C.; Spencer, P.; Ye, Q.N.; Camarda, K.V. Computational molecular design of water compatible dentin adhesive system. Comput. Aided Chem. Eng. 2015, 37, 2081–2086. [Google Scholar] [CrossRef]
- Pielichowski, K.; Bogdał, D.; Pielichowski, J.; Boroń, A. Thermal decomposition of the copolymers based on long-chained diol dimethacrylates and BIS-GMA/TEGDMA. Thermochim. Acta 1997, 307, 155–165. [Google Scholar] [CrossRef]
- Achilias, S.; Karabela, M.; Sideridou, I. Thermal degradation of light-cured dimethacrylate resins. Part, I. Isoconversional kinetic analysis. Thermochim. Acta 2008, 472, 74–83. [Google Scholar] [CrossRef]
- Achilias, S.; Karabela, M.; Sideridou, I. Thermal degradation and isoconversional kinetic analysis of light-cured dimethacrylate copolymers. J. Therm. Anal. Calorim. 2010, 99, 917–923. [Google Scholar] [CrossRef]
- Kumari, C.M.; Bhat, K.M.; Bansal, R. Evaluation of surface roughness of different restorative composites after polishing using atomic force microscopy. J. Conserv. Dent. 2016, 19, 56–62. [Google Scholar] [CrossRef]
- Janus, J. Surface roughness and morphology of three nanocomposites after two different polishing treatments by a multitechnique approach. Dent. Mater. 2010, 26, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Grzywna, Z.J.; Krasowska, M.; Ostrowski, L.; Stolarczyk, J. Can generalized dimension (Dq) and f(a) be used in structure—Morphology analysis? Acta Phys. Pol. B 2001, 32, 1561–1578. [Google Scholar]
- Krasowska, M.; Strzelewicz, A.; Dudek, G.; Rybak, A.; Barszczewska-Rybarek, I.; Turczyn, R. Fractal geometry characterization of fracture profiles of polymeric materials. A Phys. Pol. B 2014, 45, 2011. [Google Scholar] [CrossRef]
- Cullity, B.D.; Stock, S.R. Elements of X-ray Diffraction, 3rd ed.; Pearson: Essex, UK, 2001; pp. 557–572. [Google Scholar]
- Michel, V. Terminology for biorelated polymers and applications (IUPAC Recommendations 2012). Pure Appl. Chem. 2012, 84, 377–410. [Google Scholar] [CrossRef]
- Bhola, R.; Bhola, S.M.; Liang, H.; Mishra, B. Biocompatible denture polymers—A review. Trends Biomater. Artif. Organs 2010, 23, 129–136. [Google Scholar]
- Monsees, T.K. Biocompatibility and anti-microbiological activity characterization of novel coatings for dental implants: A primer for non-biologists. Front. Mater. 2016, 3, 40. [Google Scholar] [CrossRef] [Green Version]
- ANSI/ADA Standard No. 41. Evaluation of Biocompatibility of Medical Devices Used in Dentistry—ADA41-2005D; American National Standards Institute/American Dental Association: Chicago, IL, USA, 2005; Available online: https://www.ada.org/en/science-research/dental-standards (accessed on 4 November 2019).
- ISO/TR 10993-1:22. Biological Evaluation of Medical Devices. International Standard Organization. Technical Committee: ISO/TC 194 Biological and Clinical Evaluation of Medical Devices; ICS: Geneva, Switzerland, 2018; Available online: www.iso.org (accessed on 4 November 2019).
- Pawłowska, E.; Loba, K.; Błasiak, J.; Szczepańska, J. Właściwości i ryzyko stosowania metakrylanu bisfenolu A i dimetakrylanu uretanu—Podstawowych monomerów kompozytów stomatologicznych. Dent. Med. Probl. 2009, 46, 477–485. [Google Scholar]
- Mousavinasab, S.M. Biocompatibility of composite resins. Dent. Res. J. (Isfahan) 2011, 8, S21–S29. [Google Scholar]
- Schmalz, G. Resin-Based Composites. In Biocompatibility of Dental Materials; Schmalz, G., Arenholt, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 99–130. [Google Scholar] [CrossRef]
- Ferracane, J.L. Elution of leachable components from composites. J. Oral. Rehabil. 1994, 21, 441–445. [Google Scholar] [CrossRef]
- Sandborgh-Englund, G.; Nygren, A.T.; Ekstrand, J.; Elinder, C.G. No evidence of renal toxicity from amalgam fillings. Am. J. Physiol. 1996, 271, R941–945. [Google Scholar] [CrossRef]
- Issa, Y.; Watts, D.C.; Brunton, P.A.; Waters, C.M.; Duxbury, A.J. Resin composite monomers alter MTT and LDH activity of human gingival fibroblasts in vitro. Dent. Mater. 2004, 20, 12–20. [Google Scholar] [CrossRef]
- Reichl, F.X.; Simon, S.; Esters, M.; Seiss, M.; Kehe, K.; Kleinsasser, N.; Hickel, R. Cytotoxicity of dental composite (co)monomers and the amalgam component Hg(2+) in human gingival fibroblasts. Arch. Toxicol. 2006, 80, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Geurtsen, W. Biocompatibility of resin-modified filling materials. Crit. Rev. Oral. Biol. Med. 2000, 11, 333–355. [Google Scholar] [CrossRef] [PubMed]
- Bakopoulou, A.; Papadopoulos, T.; Garefis, P. Molecular toxicology of substances released from resin–based dental restorative materials. Int. J. Mol. Sci. 2009, 10, 3861–3899. [Google Scholar] [CrossRef] [PubMed]
- Moharamzadeh, K.; Brook, I.M.; Van Noort, R. Biocompatibility of resin-based dental materials. Materials 2009, 2, 514–548. [Google Scholar] [CrossRef] [Green Version]
- Pulgar, R.; Olea-Serrano, M.F.; Novillo-Fertrell, A.; Rivas, A.; Pazos, P.; Pedraza, V.; Navajas, J.M.; Olea, N. Determination of bisphenol A and related aromatic compounds released from Bis- GMA based composites and sealants by high performance liquid chromatography. Environ. Health Perspect. 2000, 108, 21–27. [Google Scholar] [CrossRef]






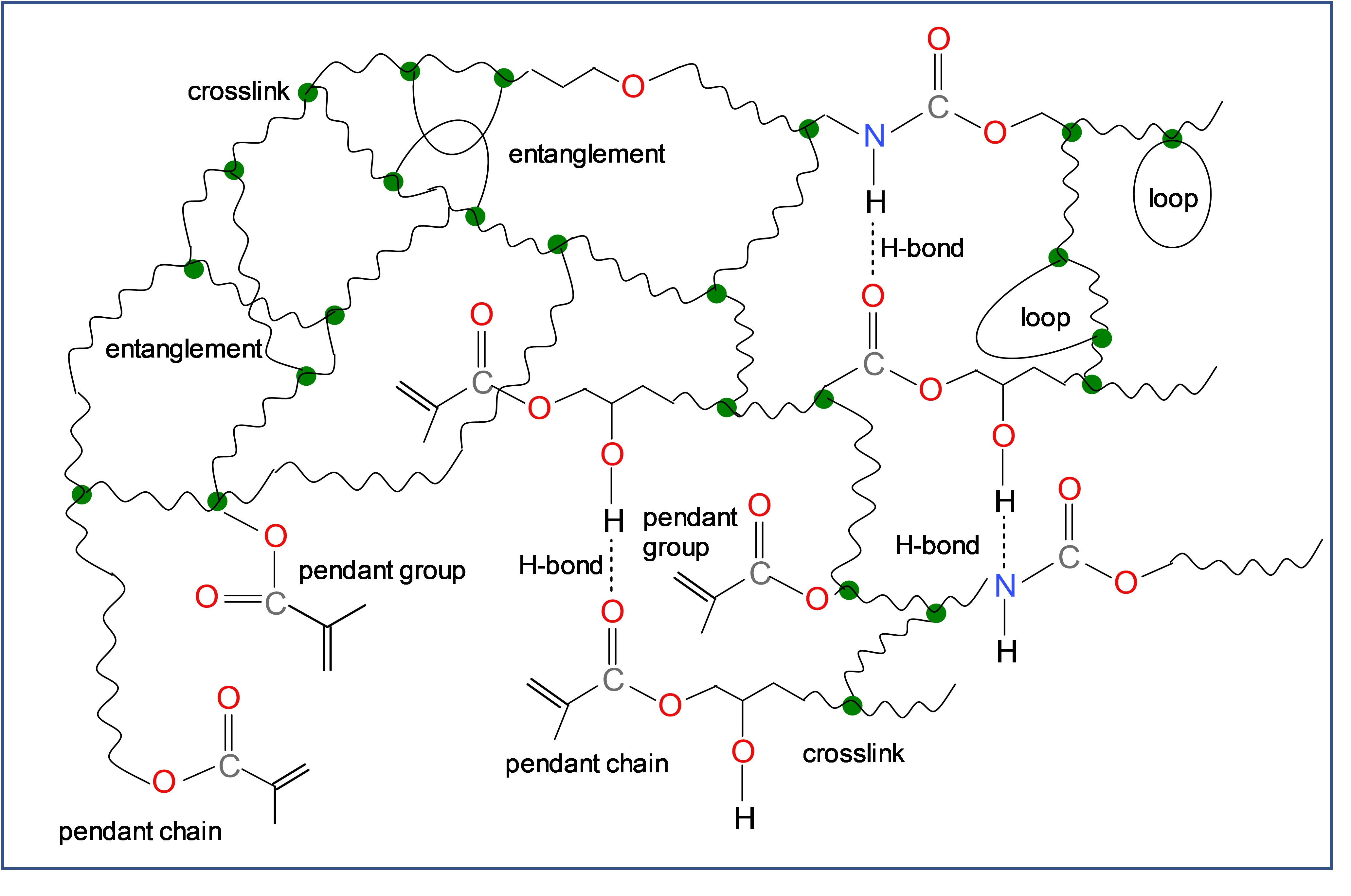{kind=link}
{kind=link}
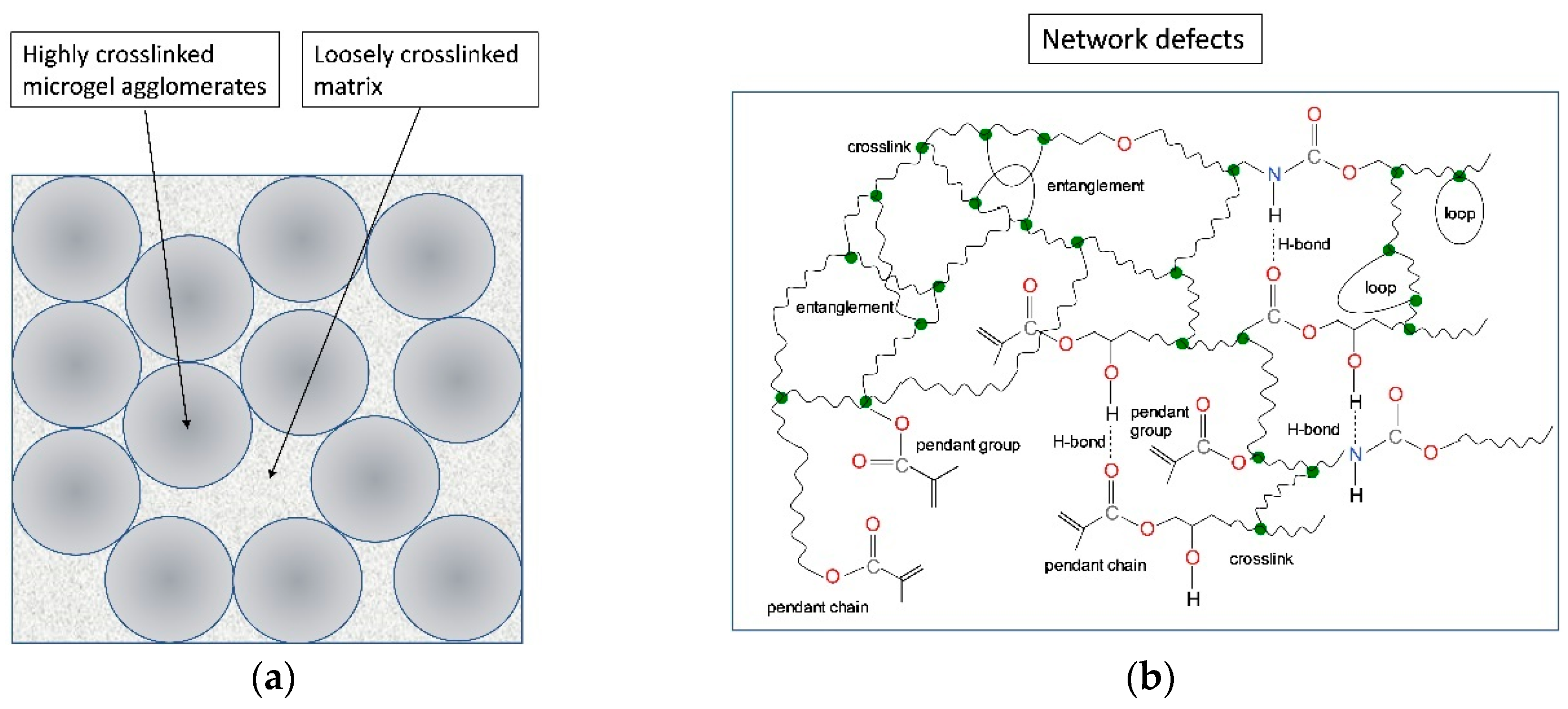
{kind=link}
{kind=link}
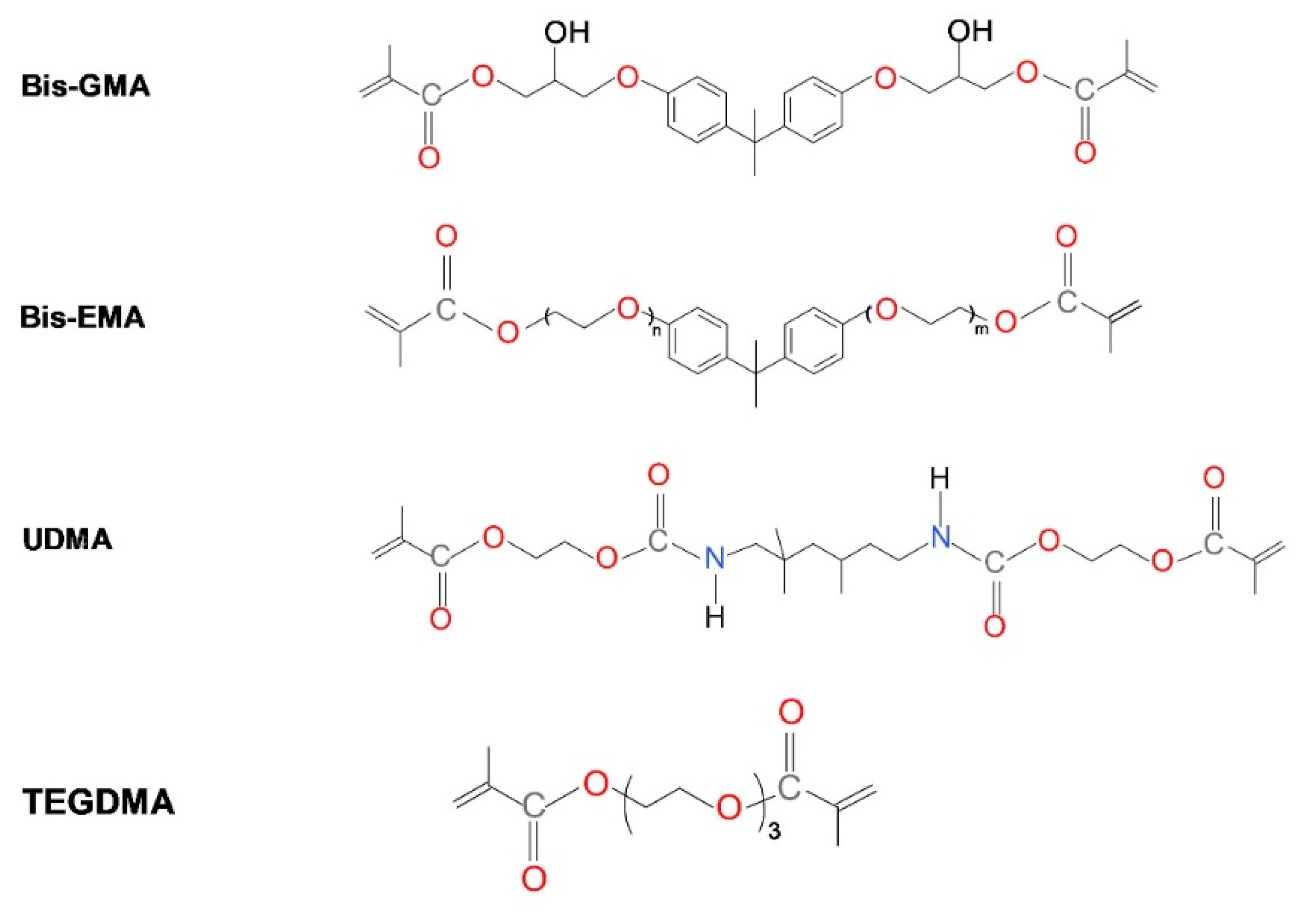
{kind=link}
{kind=link}
| Monomer | Molecular Weight (g/mol) | Concentration of Double Bonds (mol/kg) | Viscosity (Pa∙s) | Degree of Conversion (%) |
|---|---|---|---|---|
| Bis-GMA | 511 | 3.90 | 1200 1 | 39.0 1/34.5 2 |
| Bis-EMA (n+m = 4) | 540 | 3.70 | 0.9 2 | 75.5 2 |
| UDMA | 470 | 4.25 | 23.1 1 | 69.6 1/72.4 2 |
| TEGDMA | 286 | 6.99 | 0.011 1 | 75.5 1/82.5 2 |
| Monomer | Water Sorption (µg/mm3) |
|---|---|
| Bis-GMA | 32.18 1, 33.49 2 |
| Bis-EMA (n = 4) | 20.10 2 |
| UDMA | 23.85 1, 29.46 2 |
| TEGDMA | 66.93 1, 69.51 2 |
| Proton Donor Group | H-Bond Type | Wavenumber (cm−1) |
| ν(N–H) | – | 3445–3450 1 |
| ν(N–H) | N–H...N–H | 3315–3340 1 |
| ν(N–H) | N–H...O | 3260–3290 1 |
| Proton Acceptor Group | H-Bond Type | Wavenumber (cm−1) |
| ν(C=O) | – | 1730–1740 1 1721–1726 2 |
| ν(C=O) | C=O...H–N | 1703–1710 1 |
| ν(C=O) | C=O...H–O | 1712–1719 2 1705 2,3 |
| The Hydrogen Bond Type | Energy (kJ/mol) |
|---|---|
| Ο−Η.......Ν | 29 |
| Ο−Η.......Ο | 21 |
| Ν−Η.......Ν | 13 |
| Ν−Η.......Ο | 8 |
| Monomer | Young’s Modulus (MPa) | Flexural Modulus (MPa) | Flexural Strength (MPa) | Hardness (MPa) | Impact Resistance (kJ/m2) |
|---|---|---|---|---|---|
| Bis-GMA | 1427 1 | 1000 2 | 72.4 2 | 73 3 | 6.41 4 |
| Bis-EMA (n = 4) | 744 1 | 1100 2 | 87.3 2 | – | – |
| UDMA | 1405 1 | 1800 2 | 133.8 2 | 162 3 | 4.85 4 |
| TEGDMA | 1134 1 | 1700 2 | 99.1 2 | 129 3 | 8.83 4 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barszczewska-Rybarek, I.M. A Guide through the Dental Dimethacrylate Polymer Network Structural Characterization and Interpretation of Physico-Mechanical Properties. Materials 2019, 12, 4057. https://0-doi-org.brum.beds.ac.uk/10.3390/ma12244057
Barszczewska-Rybarek IM. A Guide through the Dental Dimethacrylate Polymer Network Structural Characterization and Interpretation of Physico-Mechanical Properties. Materials. 2019; 12(24):4057. https://0-doi-org.brum.beds.ac.uk/10.3390/ma12244057
Chicago/Turabian StyleBarszczewska-Rybarek, Izabela Maria. 2019. "A Guide through the Dental Dimethacrylate Polymer Network Structural Characterization and Interpretation of Physico-Mechanical Properties" Materials 12, no. 24: 4057. https://0-doi-org.brum.beds.ac.uk/10.3390/ma12244057