
Kalkitoxin Inhibits Angiogenesis, Disrupts Cellular Hypoxic Signaling, and Blocks Mitochondrial Electron Transport in Tumor Cells

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
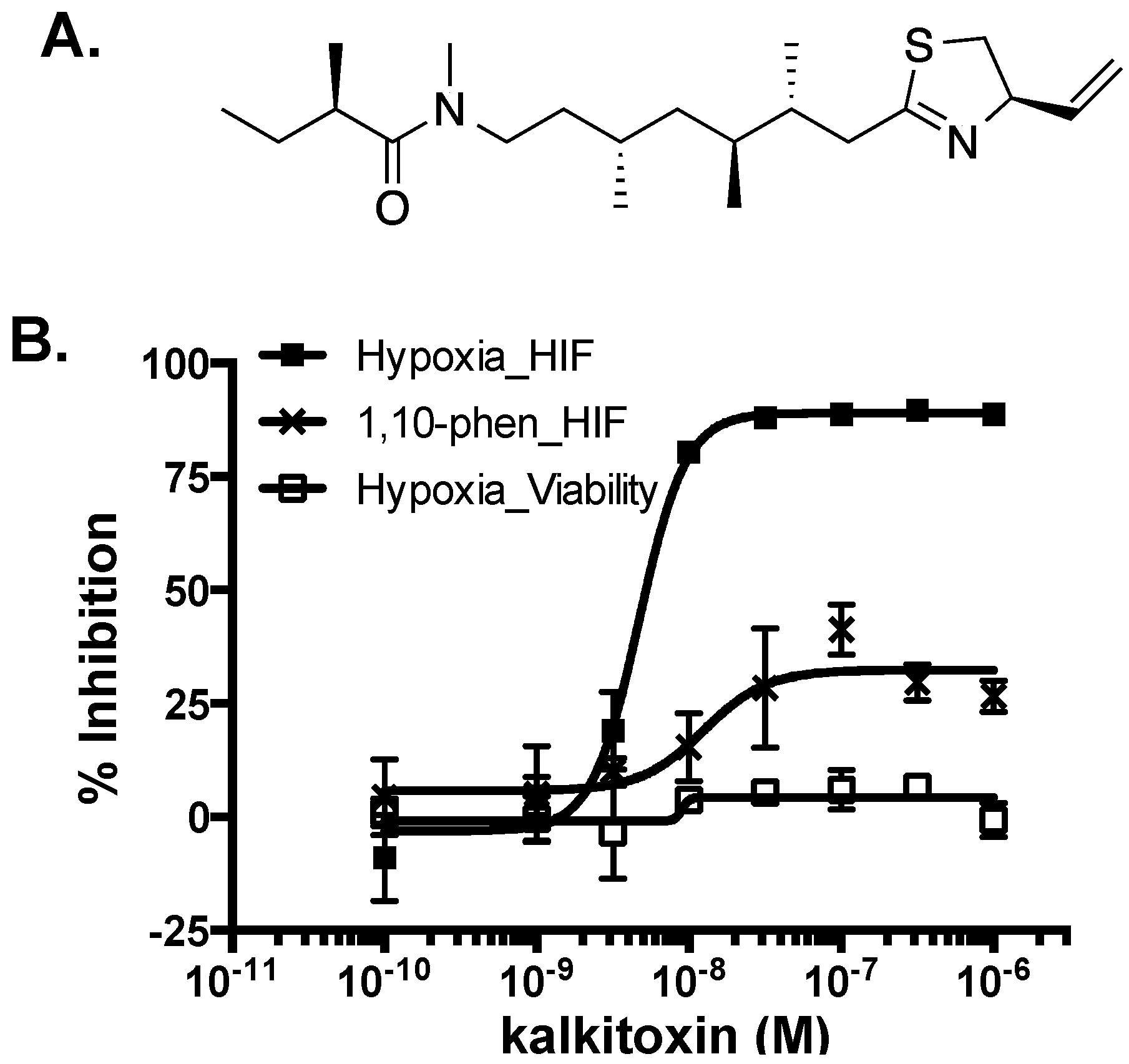
2.1. HIF-1 Inhibitory Activity

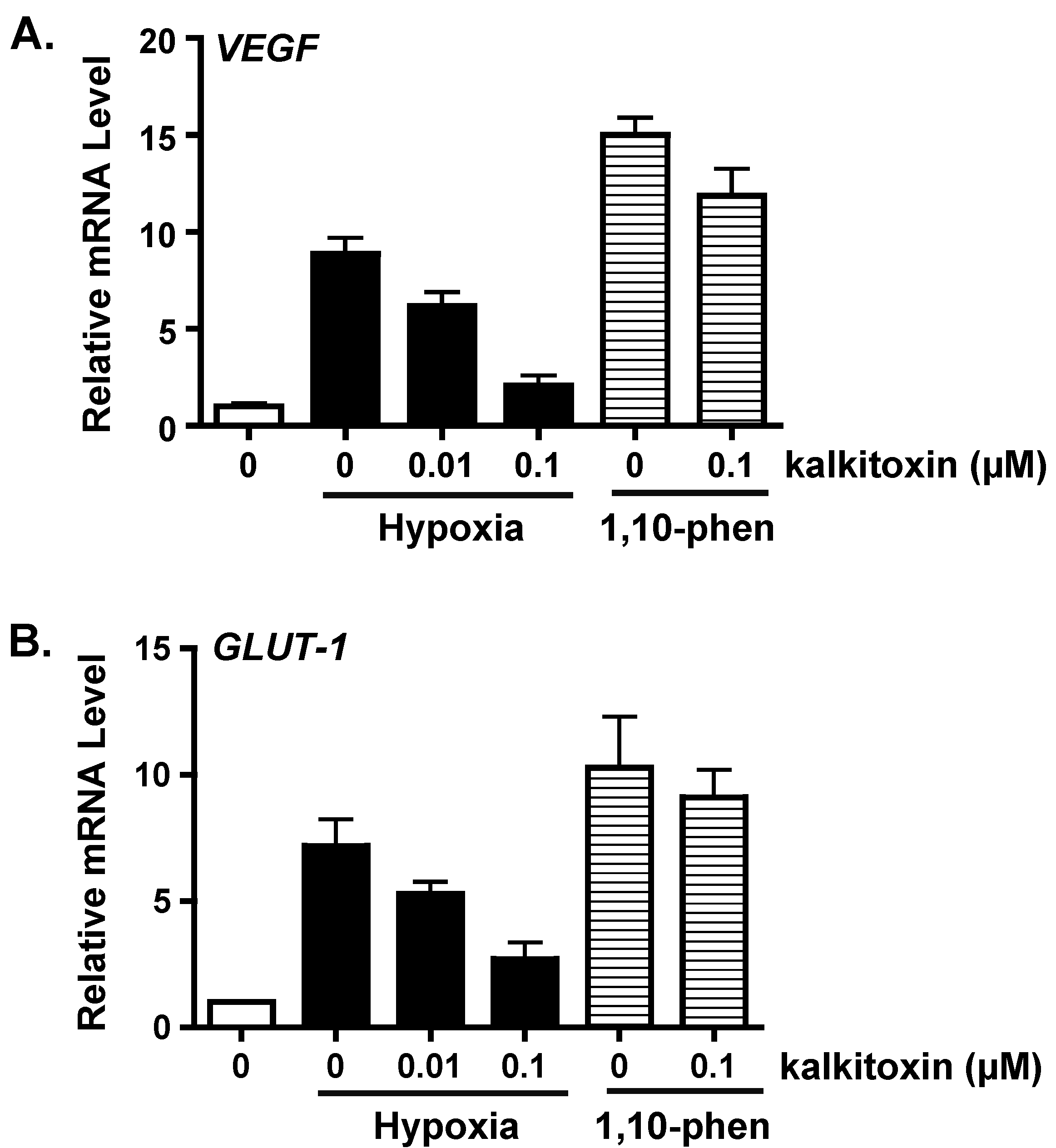
2.2. Suppression of HIF-1 Target Gene Expression

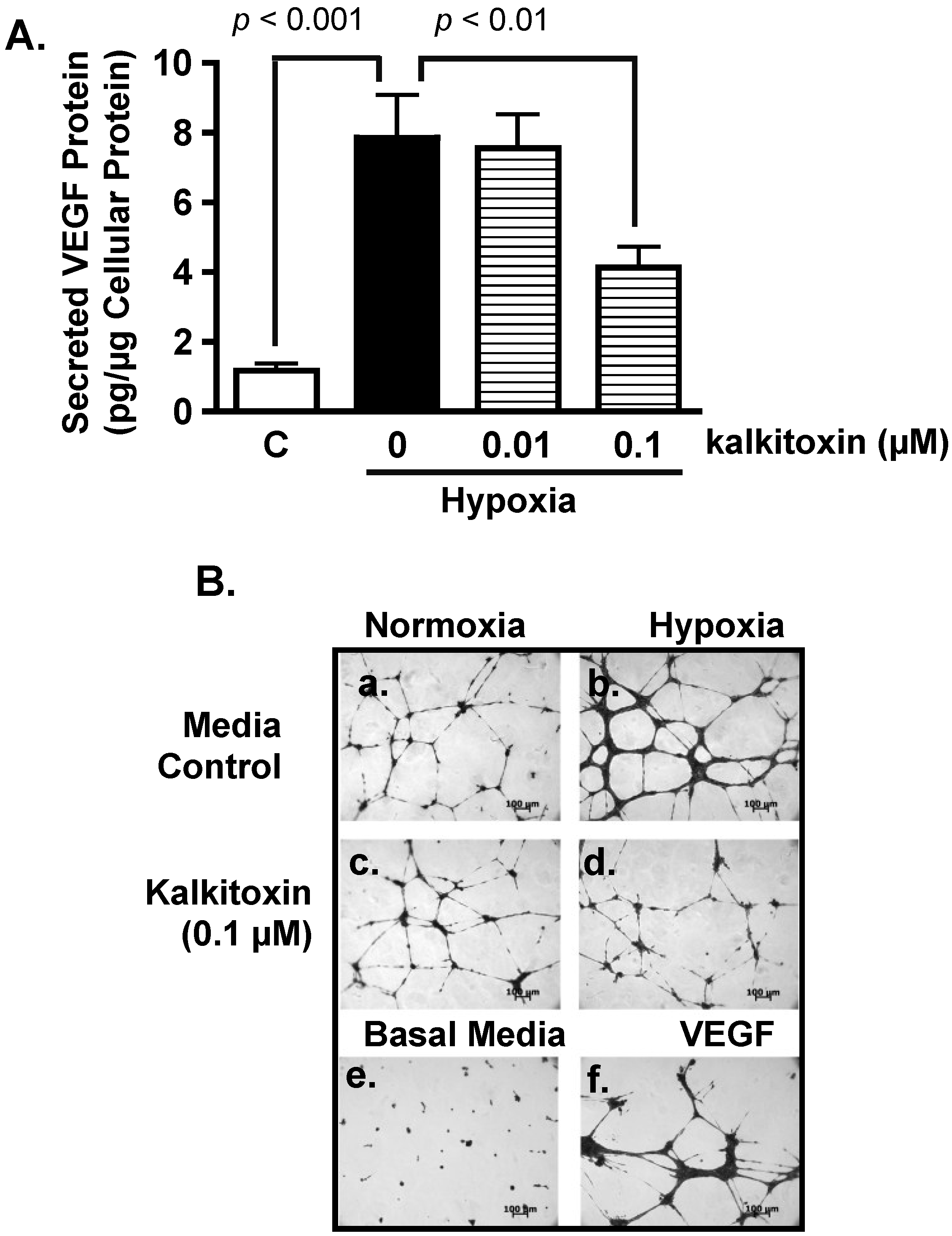
2.3. Inhibition of Hypoxia-Induced Angiogenesis


2.4. Mechanism of Action Studies
2.4.1. HIF-1α Expression

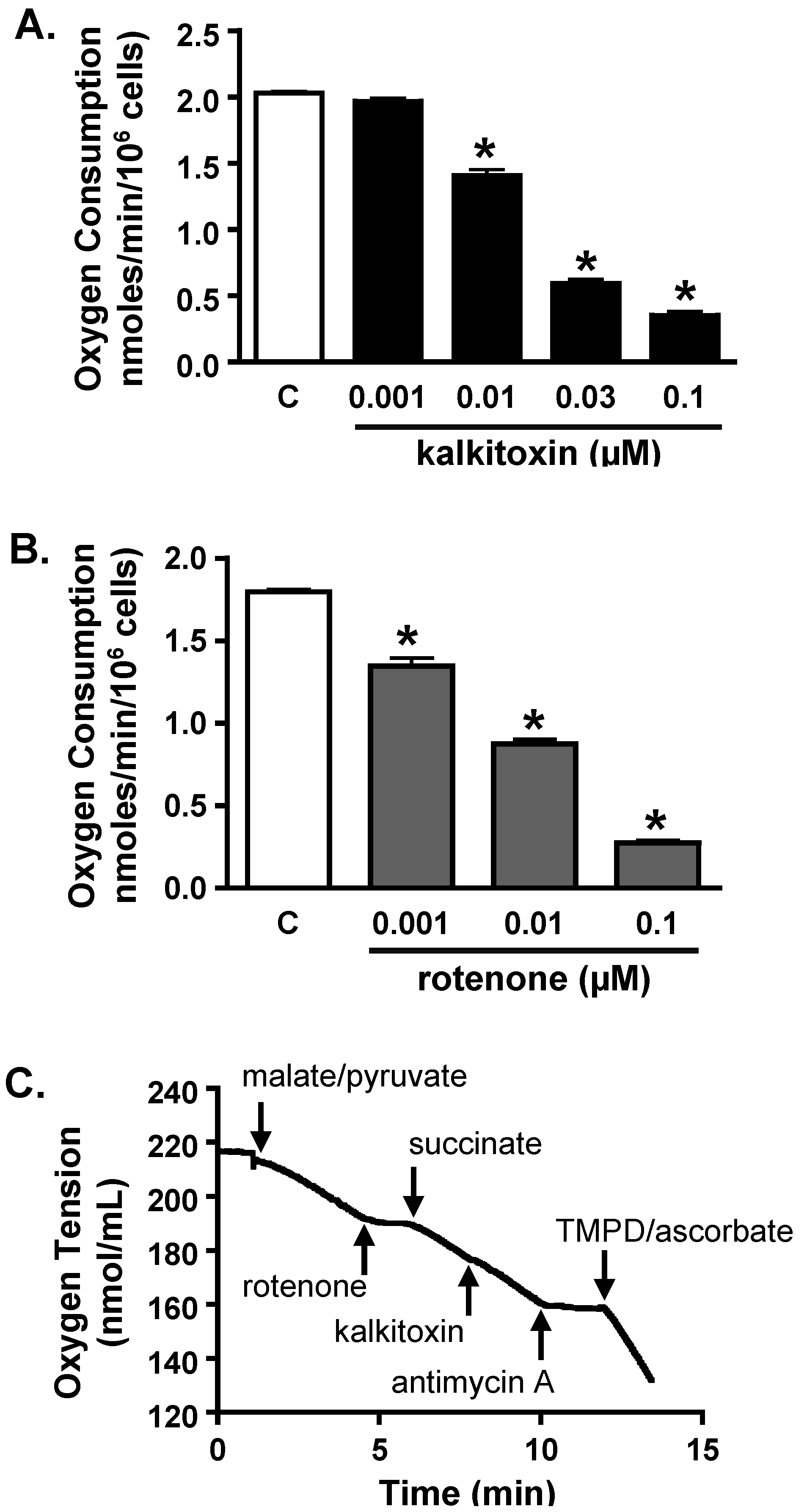
2.4.2. Mitochondrial Respiration Studies


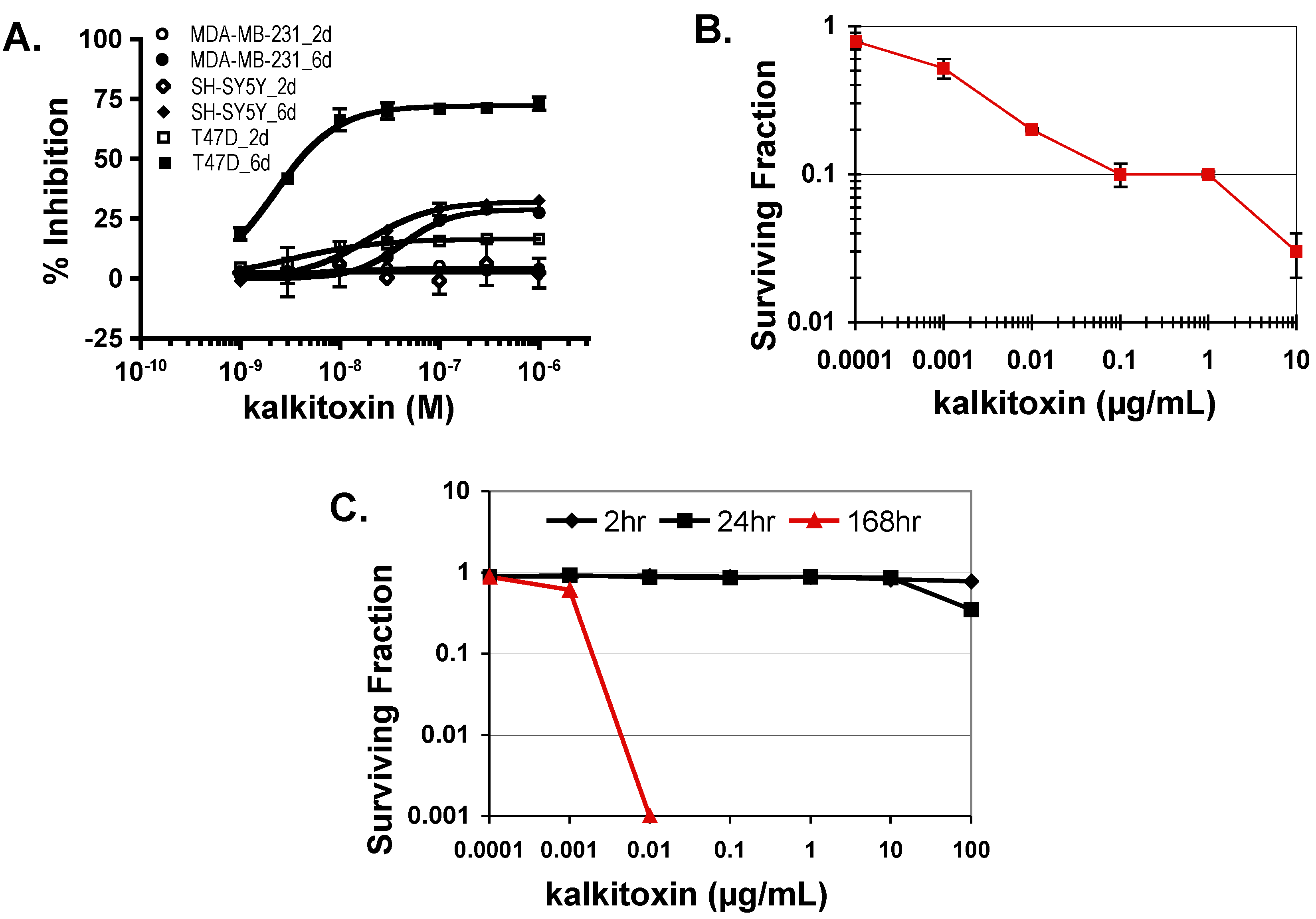
2.5. Tumor Cell Proliferation/Viability

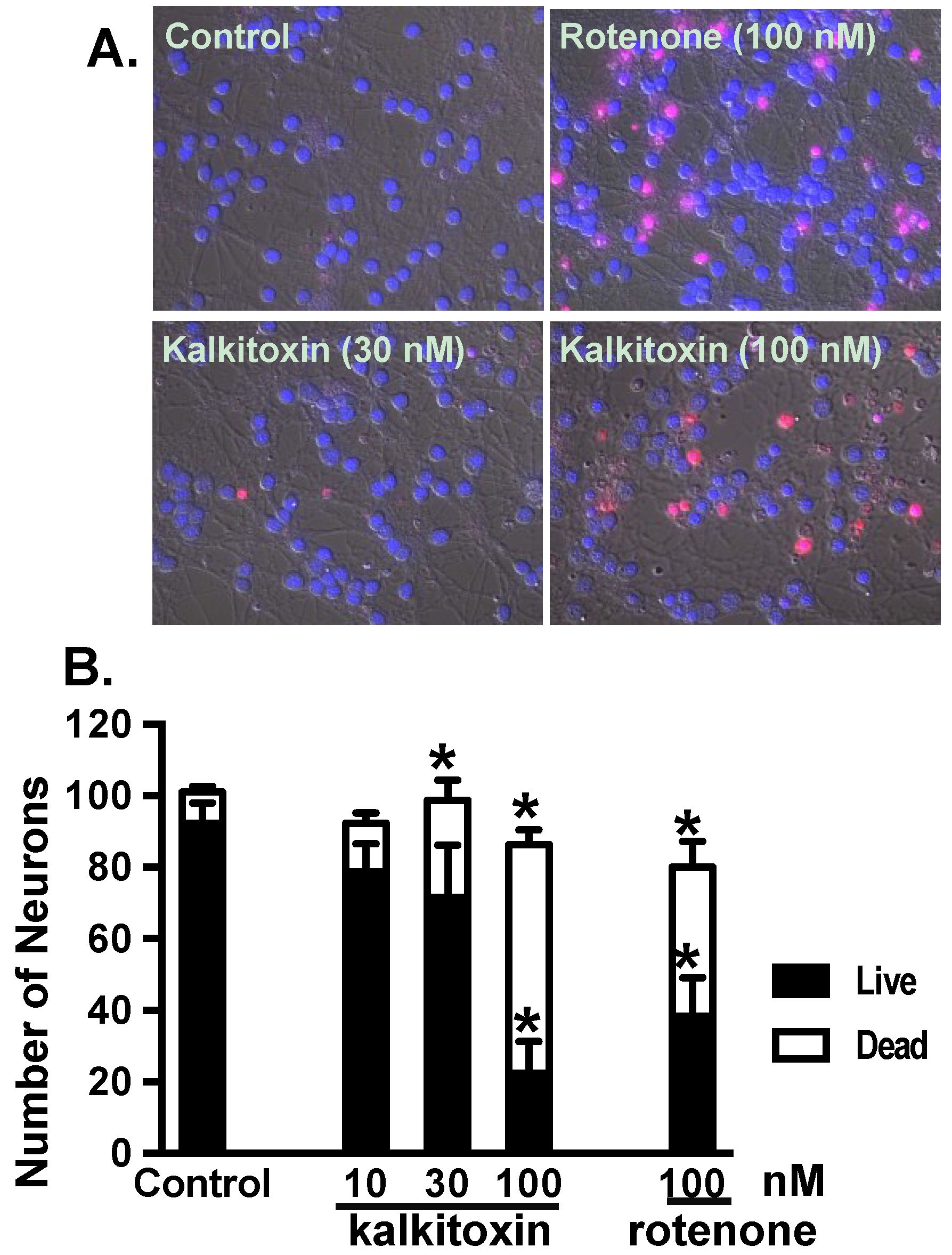
2.6. Neurotoxicity

3. Experimental Section
3.1. Tumor Cell Culture, Cell-Based Reporter and Viability Assays
3.2. RNA Extraction and Quantitative Real Time RT-PCR
3.3. ELISA Assay for Human VEGF Protein
3.4. HUVEC-Based Tube Formation Assay
3.5. Nuclear Extract Preparation and Western Blot Analysis
3.6. Mitochondria Respiration Assay
3.7. Cerebellar Granule Neuron Preparation and Neurotoxicity Assay
3.8. Statistical Analysis
4. Conclusions

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Engene, N.; Rottacker, E.C.; Kastovsky, J.; Byrum, T.; Choi, H.; Ellisman, M.H.; Komarek, J.; Gerwick, W.H. Moorea producens gen. nov., sp. nov. and Moorea bouillonii comb. nov., tropical marine cyanobacteria rich in bioactive secondary metabolites. Int. J. Syst. Evol. Microbiol. 2012, 62, 1171–1178. [Google Scholar]
- Osborne, N.J.; Shaw, G.R.; Webb, P.M. Health effects of recreational exposure to Moreton Bay, Australia waters during a Lyngbya majuscula bloom. Environ. Int. 2007, 33, 309–314. [Google Scholar]
- Albert, S.; O’Neil, J.M.; Udy, J.W.; Ahern, K.S.; O’Sullivan, C.M.; Dennison, W.C. Blooms of the cyanobacterium Lyngbya majuscula in coastal Queensland, Australia: Disparate sites, common factors. Mar. Pollut. Bull. 2005, 51, 428–437. [Google Scholar]
- Sharp, K.; Arthur, K.E.; Gu, L.; Ross, C.; Harrison, G.; Gunasekera, S.P.; Meickle, T.; Matthew, S.; Luesch, H.; Thacker, R.W.; et al. Phylogenetic and chemical diversity of three chemotypes of bloom-forming Lyngbya species (Cyanobacteria: Oscillatoriales) from reefs of southeastern Florida. Appl. Environ. Microbiol. 2009, 75, 2879–2888. [Google Scholar]
- Leao, P.N.; Engene, N.; Antunes, A.; Gerwick, W.H.; Vasconcelos, V. The chemical ecology of cyanobacteria. Nat. Prod. Rep. 2012, 29, 372–391. [Google Scholar]
- Nunnery, J.K.; Mevers, E.; Gerwick, W.H. Biologically active secondary metabolites from marine cyanobacteria. Curr. Opin. Biotechnol. 2010, 21, 787–793. [Google Scholar]
- Wu, M. Novel Bioactive Secondary Metabolites from the Marine Cyanobacterium Lyngbya majuscula. Master Thesis, Oregon State University, Corvallis, OR, USA, 1996. [Google Scholar]
- Wu, M.; Okino, T.; Nogle, L.M.; Marquez, B.L.; Williamson, R.T.; Sitachitta, N.; Berman, F.W.; Murray, T.F.; McGough, K.; Jacobs, R.; et al. Structure, synthesis, and biological properties of kalkitoxin, a novel neurotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2000, 122, 12041–12042. [Google Scholar]
- Umezawa, T.; Sueda, M.; Kamura, T.; Kawahara, T.; Han, X.; Okino, T.; Matsuda, F. Synthesis and biological activity of kalkitoxin and its analogues. J. Org. Chem. 2012, 77, 357–370. [Google Scholar]
- Berman, F.W.; Gerwick, W.H.; Murray, T.F. Antillatoxin and kalkitoxin, ichthyotoxins from the tropical cyanobacterium Lyngbya majuscula, induce distinct temporal patterns of NMDA receptor-mediated neurotoxicity. Toxicon 1999, 37, 1645–1648. [Google Scholar]
- LePage, K.T.; Goeger, D.; Yokokawa, F.; Asano, T.; Shioiri, T.; Gerwick, W.H.; Murray, T.F. The neurotoxic lipopeptide kalkitoxin interacts with voltage-sensitive sodium channels in cerebellar granule neurons. Toxicol Lett. 2005, 158, 133–139. [Google Scholar]
- White, J.D.; Xu, Q.; Lee, C.S.; Valeriote, F.A. Total synthesis and biological evaluation of +-kalkitoxin, a cytotoxic metabolite of the cyanobacterium Lyngbya majuscula. Org. Biomol. Chem. 2004, 2, 2092–2102. [Google Scholar]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar]
- Meijer, T.W.; Kaanders, J.H.; Span, P.N.; Bussink, J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin. Cancer. Res. 2012, 18, 5585–5594. [Google Scholar]
- Warfel, N.A.; El-Deiry, W.S. HIF-1 signaling in drug resistance to chemotherapy. Curr. Med. Chem. 2014, 21, 3021–3028. [Google Scholar]
- Hodges, T.W.; Hossain, C.F.; Kim, Y.P.; Zhou, Y.-D.; Nagle, D.G. Molecular-targeted antitumor agents: The Saururus cernuus dineolignans manassantin B and 4-O-demethylmanassantin B are potent inhibitors of hypoxia-activated HIF-1. J. Nat. Prod. 2004, 67, 767–771. [Google Scholar]
- Du, L.; Mahdi, F.; Datta, S.; Jekabsons, M.B.; Zhou, Y.-D.; Nagle, D.G. Structures and mechanisms of antitumor agents: xestoquinones uncouple cellular respiration and disrupt HIF signaling in human breast tumor cells. J. Nat. Prod. 2012, 75, 1553–1559. [Google Scholar]
- Li, J.; Du, L.; Kelly, M.; Zhou, Y.-D.; Nagle, D.G. Structures and potential antitumor activity of sesterterpenes from the marine sponge Hyrtios communis. J. Nat. Prod. 2013, 76, 1492–1497. [Google Scholar]
- Du, L.; Zhou, Y.-D.; Nagle, D.G. Inducers of hypoxic response: marine sesquiterpene quinones activate HIF-1. J. Nat. Prod. 2013, 76, 1175–1181. [Google Scholar]
- Schindl, M.; Schoppmann, S.F.; Samonigg, H.; Hausmaninger, H.; Kwasny, W.; Gnant, M.; Jakesz, R.; Kubista, E.; Birner, P.; Oberhuber, G. Overexpression of hypoxia-inducible factor 1α is associated with an unfavorable prognosis in lymph node-positive breast cancer. Austrian Breast and Colorectal Cancer Study Group. Clin. Cancer Res. 2002, 8, 1831–1837. [Google Scholar]
- Gong, L.; Zhang, W.; Zhou, J.; Lu, J.; Xiong, H.; Shi, X.; Chen, J. Prognostic value of HIFs expression in head and neck cancer: a systematic review. PLoS One 2013, 8, e75094. [Google Scholar]
- Shaida, N.; Chan, P.; Turley, H.; Jones, C.M.; Kanga, S.; Ritchie, R.W.; Malone, P.R.; Harris, A.L.; Fox, S.B. Nuclear localization of factor inhibitor hypoxia-inducible factor in prostate cancer is associated with poor prognosis. J. Urol. 2011, 185, 1513–1518. [Google Scholar]
- Ferrara, N.; Mass, R.D.; Campa, C.; Kim, R. Targeting VEGF-A to treat cancer and age-related macular degeneration. Annu. Rev. Med. 2007, 58, 491–504. [Google Scholar]
- Liu, Y.; Veena, C.K.; Morgan, J.B.; Mohammed, K.A.; Jekabsons, M.B.; Nagle, D.G.; Zhou, Y.-D. Methylalpinumisoflavone inhibits hypoxia-inducible factor-1 (HIF-1) activation by simultaneously targeting multiple pathways. J. Biol. Chem. 2009, 284, 5859–5868. [Google Scholar]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar]
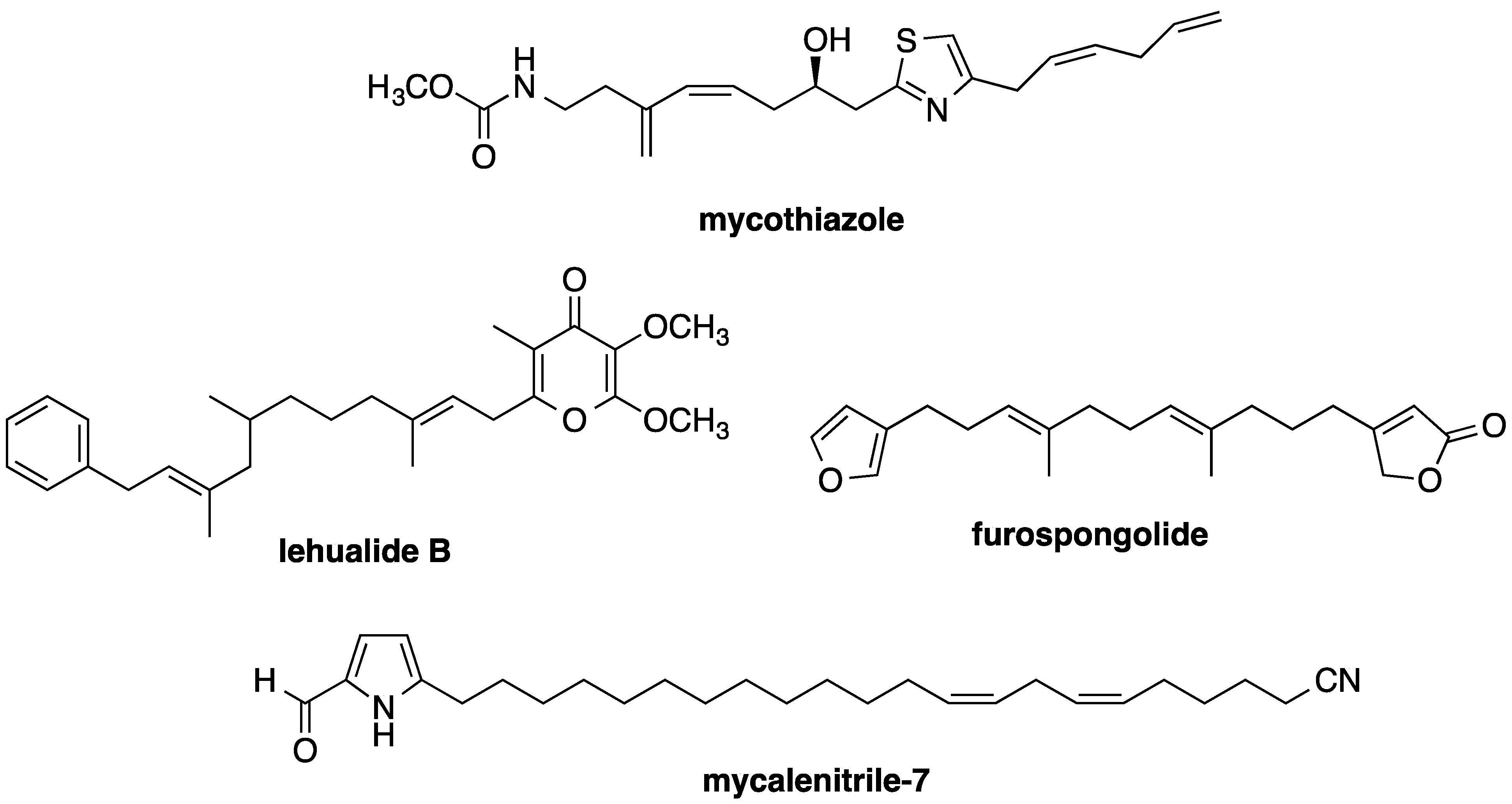
- Morgan, J.B.; Mahdi, F.; Liu, Y.; Coothankandaswamy, V.; Jekabsons, M.B.; Johnson, T.A.; Sashidhara, K.V.; Crews, P.; Nagle, D.G.; Zhou, Y.-D. The marine sponge metabolite mycothiazole: A novel prototype mitochondrial complex I inhibitor. Bioorg. Med. Chem. 2010, 18, 5988–5994. [Google Scholar]
- Coothankandaswamy, V.; Liu, Y.; Mao, S.C.; Morgan, J.B.; Mahdi, F.; Jekabsons, M.B.; Nagle, D.G.; Zhou, Y.-D. The alternative medicine pawpaw and its acetogenin constituents suppress tumor angiogenesis via the HIF-1/VEGF pathway. J. Nat. Prod. 2010, 73, 956–961. [Google Scholar]
- Li, J.; Mahdi, F.; Du, L.; Datta, S.; Nagle, D.G.; Zhou, Y.-D. Mitochondrial respiration inhibitors suppress protein translation and hypoxic signaling via the hyperphosphorylation and inactivation of translation initiation factor eIF2α and elongation factor eEF2. J. Nat. Prod. 2011, 74, 1894–1901. [Google Scholar]
- Developmental Therapeutics Program NCI/NIH. Screening Services: NCI-60 DTP Human Tumor Cell Line Screen. Available online: http://dtp.nci.nih.gov/branches/btb/ivclsp.html (accessed on 5 January 2015).
- McLaughlin, J.L. Paw paw and cancer: Annonaceous acetogenins from discovery to commercial products. J. Nat. Prod. 2008, 71, 1311–1321. [Google Scholar]
- Ayala, A.; Venero, J.L.; Cano, J.; Machado, A. Mitochondrial toxins and neurodegenerative diseases. Front. Biosci. 2007, 12, 986–1007. [Google Scholar]
- Hollerhage, M.; Matusch, A.; Champy, P.; Lombes, A.; Ruberg, M.; Oertel, W.H.; Hoglinger, G.U. Natural lipophilic inhibitors of mitochondrial complex I are candidate toxins for sporadic neurodegenerative tau pathologies. Exp. Neurol. 2009, 220, 133–142. [Google Scholar]
- Valeriote, F.A.; Tenney, K.; Medla, J.; Pietraszkiewicz, H.; Edelstein, M.; Johnson, T.A.; Taro Amagata, T.; Crews, P. Discovery and development of anticancer agents from marine sponges: perspectives based on a chemistry-experimental therapeutics collaborative program. J. Exp. Ther. Oncol. 2012, 10, 119–134. [Google Scholar]
- Jeso, V.; Yang, C.; Cameron, M.D.; Cleveland, J.L.; Micalizio, G.C. Synthesis and SAR of lehualide B—A marine-derived natural product with potent anti-multiple myeloma activity. ACS Chem. Biol. 2013, 8, 1241–1252. [Google Scholar]
- Liu, Y.; Liu, R.; Mao, S.-C.; Morgan, J.B.; Jekabsons, M.B.; Zhou, Y.-D.; Nagle, D.G. Molecular-targeted antitumor agents 19: furospongolide from a marine Lendenfeldia sp. sponge inhibits hypoxia-inducible factor-1 (HIF-1) activation in breast tumor cells. J. Nat. Prod. 2008, 71, 1854–1860. [Google Scholar]
- Mao, S.-C.; Liu, Y.; Morgan, J.B.; Jekabsons, M.B.; Zhou, Y.-D.; Nagle, D.G. Lipophilic 2,5-disubstituted pyrroles from the marine sponge Mycale sp. inhibit mitochondrial respiration and HIF-1 activation. J. Nat. Prod. 2009, 72, 1927–1936. [Google Scholar]
- Liu, Y.; Morgan, J.B.; Coothankandaswamy, V.; Liu, R.; Jekabsons, M.B.; Mahdi, F.; Nagle, D.G.; Zhou, Y.-D. The Caulerpa pigment caulerpin inhibits HIF-1 activation and mitochondrial respiration. J. Nat. Prod. 2009, 72, 2104–2109. [Google Scholar]
- Mahdi, F.; Falkenberg, M.; Ioannou, E.; Roussis, V.; Zhou, Y.-D.; Nagle, D.G. Thyrsiferol inhibits mitochondrial respiration and HIF-1 activation. Phytochem. Lett. 2011, 4, 75–78. [Google Scholar]
- Anderson, P.A.V.; Roberts-Misterly, J.; Greenberg, R.M. The evolution of voltage-gated sodium channels: Were algal toxins involved? Harmful Algae 2005, 4, 95–107. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, J.B.; Liu, Y.; Coothankandaswamy, V.; Mahdi, F.; Jekabsons, M.B.; Gerwick, W.H.; Valeriote, F.A.; Zhou, Y.-D.; Nagle, D.G. Kalkitoxin Inhibits Angiogenesis, Disrupts Cellular Hypoxic Signaling, and Blocks Mitochondrial Electron Transport in Tumor Cells. Mar. Drugs 2015, 13, 1552-1568. https://0-doi-org.brum.beds.ac.uk/10.3390/md13031552
Morgan JB, Liu Y, Coothankandaswamy V, Mahdi F, Jekabsons MB, Gerwick WH, Valeriote FA, Zhou Y-D, Nagle DG. Kalkitoxin Inhibits Angiogenesis, Disrupts Cellular Hypoxic Signaling, and Blocks Mitochondrial Electron Transport in Tumor Cells. Marine Drugs. 2015; 13(3):1552-1568. https://0-doi-org.brum.beds.ac.uk/10.3390/md13031552
Chicago/Turabian StyleMorgan, J. Brian, Yang Liu, Veena Coothankandaswamy, Fakhri Mahdi, Mika B. Jekabsons, William H. Gerwick, Frederick A. Valeriote, Yu-Dong Zhou, and Dale G. Nagle. 2015. "Kalkitoxin Inhibits Angiogenesis, Disrupts Cellular Hypoxic Signaling, and Blocks Mitochondrial Electron Transport in Tumor Cells" Marine Drugs 13, no. 3: 1552-1568. https://0-doi-org.brum.beds.ac.uk/10.3390/md13031552