Application of Prodrugs to Inflammatory Diseases of the Gut

Center for Oral Health Research, College of Dentistry and Department of Internal Medicine, University of Kentucky, Medical Center, 800 Rose Street, Lexington, KY 40536, USA
*
Author to whom correspondence should be addressed.
Molecules 2008, 13(2), 452-474; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13020452
Submission received: 26 January 2008
/
Revised: 20 February 2008
/
Accepted: 21 February 2008
/
Published: 27 February 2008
(This article belongs to the Special Issue Polysaccharides: Organic Chemistry, Bioactivity and Analysis, including Those from Medicinal Plants and Fungi)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Oral delivery is the most common and preferred route of drug administration although the digestive tract exhibits several obstacles to drug delivery including motility and intraluminal pH profiles. The gut milieu represents the largest mucosal surface exposed to microorganisms with 1010-12 colony forming bacteria/g of colonic content. Approximately, one third of fecal dry matter is made of bacteria/ bacterial components. Indeed, the normal gut microbiota is responsible for healthy digestion of dietary fibers (polysaccharides) and fermentation of short chain fatty acids such as acetate and butyrate that provide carbon sources (fuel) for these bacteria. Inflammatory bowel disease (IBD) results in breakage of the mucosal barrier, an altered microbiota and dysregulated gut immunity. Prodrugs that are chemically constructed to target colonic release or are degraded specifically by colonic bacteria, can be useful in the treatment of IBD. This review describes the progress in digestive tract prodrug design and delivery in light of gut metabolic activities.
Introduction
Oral delivery is the most common and preferred route of drug administration. This is the ideal route to deliver compounds to colonic sites to treat Inflammatory Bowel Disease (IBD); however; the digestive tract exhibits several obstacles to drug delivery including gut motility [1,2], stomach and intraluminal pH profiles, and degradative enzymes [3]. Therefore, prodrugs are designed to transport intact drug through the stomach and small intestine, but be hydrolyzed by the colonic microbiota to release the active compound.
The concept of “prodrug” was first introduced by Adrien Albert in 1958 [4] to describe compounds that undergo biotransformation prior to eliciting their pharmacological effect, i.e. "therapeutic agents that are inactive but transform into one or more active metabolites." Prodrugs are designed to overcome pharmaceutical, pharmacokinetic, or pharmacodynamic barriers such as insufficient chemical stability, poor solubility, unacceptable taste or odor, irritation or pain, insufficient oral absorption, inadequate blood-brain barrier permeability, marked pre-systemic metabolism, and toxicity [5]. Furthermore, by attachment of a pro-moiety to the active moiety, a prodrug is formed which is designed to overcome the barriers that hamper the optimal use of the active principle [6].
The prodrug approach, in which a derivative of the active compound is synthesized, has been proven to be an effective and important way of overcoming barriers that restrict the application of many chemical entities as orally administered drugs [7,8]. The colonic drug delivery systems are mainly based on degradation of a prodrug by microorganisms of the colon flora [9,10] and colonic enzymes [11,12]. In this review different approaches to colonic drug delivery are discussed.
Inflammatory Bowel Disease
IBD refers to idiopathic inflammatory diseases of the intestine, principally ulcerative colitis and Crohn’s disease [13]. IBD is characterized by chronic inflammation in the mucosal membrane of the small and/or large intestine. Although many treatments have been recommended for IBD, they do not treat the cause but are effective only in reducing the inflammation and accompanying symptoms in up to 80% of patients. Thus, current medical management of IBD consists of anti-inflammatory and immunosuppressive agents and biologic drugs, as well as surgery [13,14,15]. The mortality rate of patients with Crohn’s disease is 40% greater than those without evidence of disease including Ulcerative disease [14]. In a retrospective, population based cohort study of mortality by medication use among IBD included 9032 patients during 1996-2002; with the exception of immunomodulators, the medications for IBD were not significantly associated with mortality among IBD patients. However, infections, respiratory diseases and digestive diseases other than IBD have been important specific cause of death in IBD patients. Despite medical intervention, many patients with IBD suffer from complications including abscesses, fistulae, intestinal obstruction, chronic blood loss, and intestinal neoplasia. Also, colorectal cancer is increased in IBD, particularly among male patients [14].
Recent research has lead to the development of several new treatment strategies that target specific biomolecules. There are multiple therapies in various stages of investigation, including biological agents such as human recombinant cytokines IL-10 [16], IL-11, IL-12 [17,18] and antisense oligonucleotides to intercellular adhesion molecule (ICAM), α4 integrin [19], and monoclonal antibodies to TNF-α [19,20] and IFN-γ with many more on the horizon. Although these approaches are exciting and represent a move forward in the treatment of IBD, several issues remain. These include cost, long-term safety, adverse effects, and the requirement for a parenteral route of administration. Finally, many IBD patients remain refractory or intolerant to current therapies.
Pathogenesis of IBD
The etiology of IBD remains unknown; however, two primary theories have been proffered focusing on either a specific persistent infectious agent [21,22] or an abnormal host immune response to ubiquitous antigens in the luminal constituents. Evidence supports that patients with IBD are genetically susceptible to this disease, and the defect targets an inability to effectively down-regulate the inflammatory response to specific antigens or luminal bacteria [23]. Genetically engineered animals develop spontaneous enterocolitis that resembles IBD [24]. In several of these models (e.g. IL-2-/-, IL-10-/- mice, HLA-B27 transgenic rats), the normal resident bacterial microbiota is necessary for the development of colitis [25,26]. These data suggest that in a genetically susceptible host, a dysregulated inflammatory/immune response to the nonpathogenic luminal commensal bacteria leads to a chronic inflammatory process. Inflammatory responses in the intestine can be induced by disorders of immunoregulation, infectious colitis, impaired circulation, or even toxic compounds.
Striking similarities exist in the clinical and morphologic manifestations among these various forms of intestinal inflammation. These similarities probably do not reflect directed target cell injury but rather innocent bystander effects due to the presence of soluble mediators of inflammation [27]. These mediators include reactive oxygen species (ROS), cytokines, arachidonic acid metabolites, and growth factors. Cytokines also elicit numerous metabolic responses including altered zinc metabolism [28], and synthesis of acute phase reactants that also are biochemical/clinical complications of IBD [29]. In addition, neutrophil and macrophage infiltration suggest the presence of chemotaxins in the mucosal tissues. Mucosal edema and hyperemia reflect the presence of soluble mediators that induce vascular permeability and vasodilatation [30].
LPS and TNF production and IBD
The gut milieu represents the largest mucosal surface exposed to microorganisms with 1010-12 colony forming bacteria/g of colonic content. The intestinal micobiota [31] represents 400–500 bacterial species, and encompasses more bacterial cells than eukaryotic cells within the human body [32]. Approximately, one third of fecal dry matter is made of bacteria/ /bacterial components. Indeed, the normal gut microbiota is responsible for healthy digestion of dietary fibers (polysaccharides) and fermentation of short chain fatty acids such as acetate, butyrate and propionate that provide carbon sources (fuel) for these bacteria, nutrient production, metabolism of dietary carcinogens, and conversion of prodrugs to active drugs. However, growing evidence suggests that the enteric microorganisms are critical components in the pathogenesis of IBD and bacterial endotoxemia has been postulated to play a key role in IBD [23,32].
Bacteroides and Clostridium species have been specifically implicated with inflammatory responses in different animal models of IBD [21,23], whereas other commensals such as Lactobacillus and Bifidobacterium species, have minimal proinflammatory capabilities and are less likely to translocate from the lumen to the internal milieu [23]. Lipopolysaccharide (LPS), a cell wall constituent of Gram-negative bacteria, is present in a high concentration in the normal colonic lumen. When exposed to LPS, or other stimuli, macrophages release a variety of mediators of inflammation, of which TNF-α is a prominent component that mediates some disparate effects of LPS challenge [33]. TNF-α is produced earlier than some other cytokines after an LPS challenge and comprises ~2% of the total secretory products made by activated macrophages [34]. In LPS-treated animals, TNF-α achieves peak serum concentrations 60-120 minutes after LPS administration. ROS generated during IBD are also important stimuli for TNF-α production. Administration of purified TNF-α to animals evokes the same metabolic response as LPS, including fever, anorexia, neutrophilia, and muscle wasting. These biochemical/clinical complications are also present in IBD [29]. In addition, endotoxemia and elevated serum concentrations of inflammatory cytokines, including TNF-α, are observed in IBD patients [35]. In these cases, injury is down-regulated by antibody to TNF-α [19,20,36].
NF-κB and IBD
As a transcriptional activator of genes encoding inflammatory cytokines and adhesion molecules, NF-κB is an important effector molecule in intestinal inflammation and also a suitable target for anti-inflammatory therapy. Mucosal inflammation in clinical IBD has been linked to the upregulated nuclear translocation of the pro-inflammatory transcription factor complex NF-κB and expression of pro-inflammatory cytokines, such as TNF-α. Multiple stimuli, including ROS, cytokines and endotoxin, cause release of IκB and permit NF-κB to enter the nucleus, bind to DNA control elements, and induce transcription of specific genes (e.g. mediators of inflammation in IBD). These include cytokines (e.g. IL-1, 6, 8, 12, and TNF-α), enzymes (e.g. inducible nitric oxide synthase), and cell adhesion molecules (e.g. E-selectin) [37]. Several established anti-inflammatory and immunosuppressive agents used in the treatment of IBD are known inhibitors of NF-κB. For instance, glucocorticoids block NF-κB activity in part by upregulating IκB protein levels [38,39]. Sulfasalazine [40] mesalamine [41] and Cyclosporin A, all immunosuppressants that target T cells, also inhibit NF-κB [42].
Reactive Oxygen Species (ROS)
Oxidant-mediated injury plays an important role in the pathophysiology of IBD. ROS upregulate NF-κB and result in further increases in the levels of oxidants and tissue injury in IBD. Inflamed gut from IBD patients, as well as IBD animal models, are rich in activated macrophages and neutrophils and these inflammatory cells generate excess amounts of ROS with subsequent increases in oxidative stress [43]. These ROS include hydroxyl radicals, superoxide anions, hydrogen peroxide, and nitric oxide. ROS are extremely unstable species due to their high reactivity and may result in lipid peroxidation and the oxidation of DNA and proteins [44]. The increased generation of highly toxic ROS exceeds the limited intestinal antioxidant defense system, thereby contributing to intestinal oxidative injury in IBD patients [45]. Excessive production of ROS has been demonstrated in circulating phagocytic and polymorphonuclear leukocytes in IBD patients using a chemiluminescence assay [46,47] and following stimulation with the bacterial chemotactic peptide N-formyl-methionyl-leucyl-phenylalanine (fMLP) [48]. Thus, oxidant-mediated injury plays an important role in the pathophysiology of IBD.
The Prodrug Approach
The primary goal of drug therapy in IBD is to reduce inflammation in the colon that generally requires frequent intake of high dose anti-inflammatory drugs. Thus, the prodrug approach continues to provide increased scrutiny as an alternative to this systemic approach. Three classes of Prodrugs, which target drug to the colon are currently used in IBD or are in experimental stages: (1) Anti-inflammatory agents (eg. azu prodrugs, 5-Aminosalicylic acid, amino acids) that can result from bacteria changing the function of prodrugs or prodrugs treating inflammation which changes the bacterial ecology; (2) Immunomodulators and Immunosuppressants; and, (3) Antioxidants.
Anti-Inflammatory Prodrugs:
5-Aminosalicylic acid
5-Aminosalicylic acid (5-ASA, 1a, Figure 1) is an effective compound to attenuate the inflammatory response in IBD while its mechanism of action is not fully understood. Thus, it usually fails to reach the colon leading to significant adverse effects such as ulcerogenic potential [49]. Therefore, a prodrug approach for colonic delivery of 5-ASA has become a rational system of drug delivery for the topical treatment of IBD. 5-ASA triggers the peroxisome proliferator-activated receptor (PPAR-γ) family of nuclear receptors, which regulate inflammation, cell proliferation, apoptosis, and metabolic function. PPAR-γ receptors are highly expressed in colonic epithelia and their expression is upregulated by the gut bacteria [50].
Figure 1.
(1a) Molecular structure of 5-Aminosalicylic acid (5-ASA); (1b) Molecular structure of Sulfasalazine (5-ASA linked to Sulfapyridine by the azo bond).
Figure 1.
(1a) Molecular structure of 5-Aminosalicylic acid (5-ASA); (1b) Molecular structure of Sulfasalazine (5-ASA linked to Sulfapyridine by the azo bond).

In order to achieve an effective colonic delivery, a drug needs to be protected from absorption and/or the environment of upper GI tract and then rapidly released into the proximal colon, which is the optimalsite for colon-targeted delivery of the drug. Colonic drug delivery through colon-specific prodrug activation may be accomplished by the utilization of the high activity of certain enzymes at the target site relative to non-target tissues, enabling for prodrug conversion to active drug [51].
The microbiota of the GI tract consists of a coexisting mixture of aerobic, facultative anaerobic, and strict anaerobic bacteria in a complex ecosystem. These bacteria produce a wide range of enzymes, such as β-glucuronidase, β-xylosidase, α-arabinosidase, β-galactosidase, nitroreductase, azoreductase, deaminase, urea hydroxylase, etc [52]. The reductive environment of the bacterial microbiota can be utilized for colon targeting. The prodrug approach has been successfully utilized with sulfasalazine (an azo prodrug 5-ASA and sulfapyridine, 1b, Figure 1) for targeting drugs to the colon [51]. The azo prodrugs are poorly absorbed in the stomach and small intestine. The azo linkage is cleaved by the azoreductases released by the colonic anaerobic bacteria to form a pair of amines [53]. The majority of side effects of sulfasalazine, eg. hepatotoxicity, hypospermia, and severe blood disorders, are due to sulfapyridine. Even though different prodrugs of 5-ASA like balsalazide, ipsalazine and olsalazine [9,10,11] have been reported, few of them have extended beyond the stage of clinical trials.
Balsalazide, a prodrug of 5-ASA and an analogue of sulfasalazine, is linked to the carrier 4-aminobenoyl β-alanine via an azo-bond and is specifically converted to 5-ASA by azo-reducing bacteria present in the colon [54]. The prodrug remains intact in the gastrointestinal tract until it reaches the colon. This agent uses a mode of drug administration to exert a local anti-inflammatory effect directly on the inflamed lining of the intestinal wall. Alternatively to colon targeting, many prodrug projects are directed toward avoiding gastrointestinal side effects. Olsalazine is a dimer of 5-ASA linked by an azo-bond to avoid the sulfapyridine moiety [9,10,11]. The slow release 5-ASA preparations (enteric-coated; eg. mesalamine, Pentasa, Asacol) have been approved by the FDA. Pentasa microgranules are coated with a semipermeable membrane of ethylcellulose with amphoionic properties. 5-ASA is slowly released over time from the granules into the intestine, and enhanced with pH above 6.0. [55]. However, the need for a safer, colonic delivery of prodrugs of 5-ASA with nontoxic carriers and non- ulcerogenic potential still remains.
Still in an exploratory stage, recently an amino acid (mutual) azo prodrug of 5-ASA was synthesized by coupling L-tryptophan with salicylic acid, for targeted drug delivery to the inflamed colonic tissue in IBD [56]. The structure of the synthesized prodrug was confirmed by elemental analysis, IR and NMR spectroscopy. In vitro kinetic studies showed negligible release of 5-ASA in HCl buffer (pH 1.2), whereas 18% was released in phosphate buffer (pH 7.4) over a period of 7 h. In rat fecal matter, 87.9% of 5-ASA was released with a half-life of 143.6 min, following first order kinetics. The azo conjugate was evaluated for its ulcerogenic potential by Rainsford's cold stress method. The ameliorating effect of the azo conjugate and therapeutic efficacy of the carrier system was evaluated in TNBS-induced experimental colitis model. The synthesized prodrug was found to be equally effective in attenuating the colitis in rats as that of sulfasalazine without the ulcerogenicity of 5-aminosalicylic acid.
Acid-regulation and short chain fatty acids
The proton pump inhibitor omeprazole utilizes the acidic environment of the stomach [57,58]. Omeprazole is a prodrug of a sulfonamide that exerts its potent ant-iulcer effects by covalently modifying cysteine residues on the luminal side of the proton pump (i.e., H+/K+-ATPase) in the oxyntic mucosa of the stomach. This prodrug only exerts its anti-secretory effect in the acidic environment of the oxyntic mucosa of the stomach.
Short chain fatty acids
Short chain fatty acids (SCFA), mainly acetate, propionate and butyrate, are produced in the colonic environment by action of bacterial fermentation to maintain the normal physiological function of the gut. The effects of SCFA include modulation of colonic muscular activity, stimulation of electrolyte and fluid uptake and enhancement of blood flow [59,60]. Of the major SCFA, butyrate appears to be a preferred oxidative substrate for colonocytes. It also acts to maintain a normal phenotype in these cells through repair of damage to DNA and inhibition of apoptosis in normal cells and its promotion in colon tumor cells in vitro [59,60,61] and in vivo rat treated with carcinogen azoxymethane [62]. IBD results in SCFA deficiency [63]. SCFA consumed directly in foods or beverages are rapidly absorbed in the fore-gut and fail to reach the colon [64]. However, acylated maize starch with butyric anhydride was shown to produce resistant starch that increased SCFA in the rat gut, apparently through bacterial release of the esterified fatty acid and fermentation of the residual starch [65].
Carnitine (β-hydroxy-γ-trimethylaminobutyrate)
Carnitine (β-hydroxy-γ-trimethylaminobutyrate) is obligatory for transport of long-chain fatty acids into mitochondria for subsequent β-oxidation [66]; it plays a critical role in energy metabolism of the tissues that derive a substantial portion of their metabolic energy from fatty acid oxidation such as heart, skeletal muscle, liver [67,68]. Butyrate, a bacterial fermentation product, is beneficial forprevention/treatment of ulcerative colitis. Butyryl-L-carnitine, a butyrate ester of carnitine, may have potential for treatment of IBD since butyryl-L-carnitine would supply both butyrate and carnitine. L-Carnitine is absorbed in the intestinal tract via the carnitine transporter OCTN2 and the amino acid transporter ATB. Loss-of-function mutations in OCTN2 may be associated with IBD, suggesting a role for carnitine in intestinal/colonic health. In contrast, ATB is upregulated in bowel inflammation [69]
Immunomodulators and Immunosuppressors
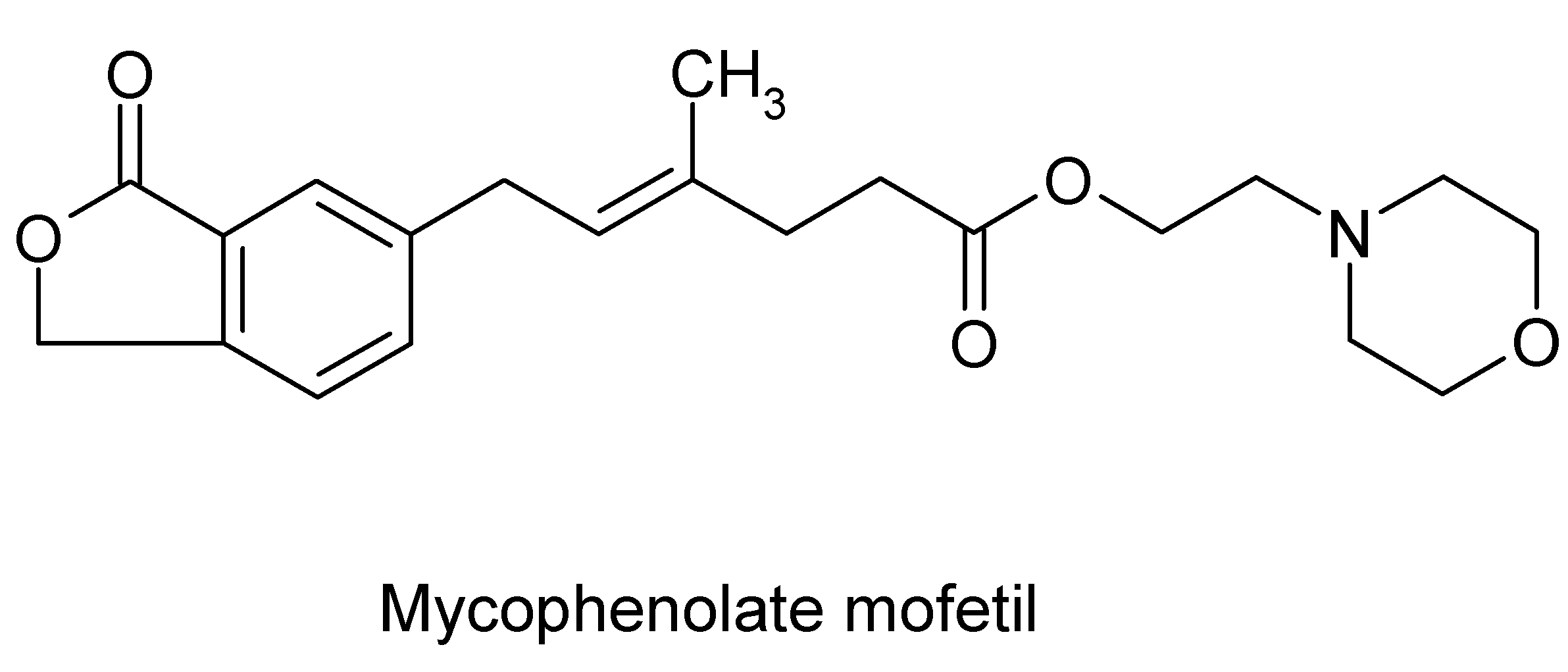
Mycophenolate mofetil
Mycophenolate mofetil (MMF, Figure 2) is an ester prodrug of mycophenolic acid, the active moiety that gives the drug its immunosuppressive properties [70]. A century ago (1896), Gosio [71] isolated mycophenolic acid from a culture Penicillium species and noted its antibacterial properties. Mycophenolic acid decreases de novo synthesis of guanosine nucleotides by reversible inhibition of inosine monophosphate dehydrogenase (IMPDH) [72]. This prodrug acts against T and B lymphocytes, because these cells depend on the de novo synthesis of purines for proliferation, whereas other cell types, such as neutrophils and macrophages, can use salvage pathways [69]. Mycophenolate mofetil is unique because of its dual activity as an immunosuppressant, as well as an antimicrobial with a novel action against Pneumocystis carinii pneumonitis [73,74,75]. Mycophenolic acid is known to inhibit growth of intestinal smooth muscle and synthesis of fibronectin [76], and may be useful to inhibit smooth-muscle hyperplasia and stricture formation in Crohn’s patients.
Figure 2.
Molecular structure of the immunomodulator mycophenolate mofetil(MMF).

A number of clinical trials [77,78,79,80] have suggested a therapeutic role for MMF in patients with ulcerative colitis and Crohn’s disease. These controlled trials have shown efficacy of MMF in patients with active IBD. Based on these trials, patients with IBD refractory to other IBD therapies (corticosteroids, azathioprine or 6-mercaptopurine, methotrexate, and infliximab, monoclonal antibody against TNFα) may be treated with MMF in selected circumstance [83]. The development of colitis in patients treated with MMF has been described in four renal-transplant recipients [81]. However, these patients were receiving high-dose MMF. The development of colitis in these patients may be caused by enterohepatic cycling of mycophenolic acid resulting in high colonic concentrations of the metabolite [82].
Azathioprine
Azathioprine, a thiopurine prodrug, is another immunosuppressant used in IBD and liver transplantation patients [77,78,80,84]. Approximately 1 in 10 patients suffers side effects in response to the drug, such as bone marrow toxicity [84]. There is evidence that polymorphisms in the genes encoding thiopurine methyltransferase and inosine triphosphate pyrophosphatase predict adverse drug reactions to Azathioprine therapy. Furthermore, common genetic polymorphisms in the gene encoding methylenetetrahydrofolate reductase may have an indirect impact on thiopurine drug methylation by influencing levels of the methyl donor S-adenosylmethionine.
Glucocorticosteroids
Corticosteroids are commonly prescribed for treatment of moderate to severe IBD however, numerous adverse effects limit their long term use [79]. Budesonide is a nonhalogenated corticosteroid with a high affinity for the glucocorticoid receptor. Budesonide is a 1:1 mixture of the epimers (22R+22S), which both are rapidly eliminated with a terminal half-life of 2.7±0.6 h [85]. Budesonide is extensively metabolized by hydroxylation, while the cytochrome P450 isoenzyme CYP3A4, expressed in high amounts in hepatocytes and epithelial cells of the intestinal wall, is the main responsible isoenzyme for its rapid elimination. “Budenofalk®” is a slow releasing Budesonide composed of a hard gelatin capsule and acid-resistant pellets. The pellets are covered with Eudragit, with a delayed release at pH > 6.4. Following ingestion of the capsules more budesonide is available in the region of the terminal ileum. [85]. Budesonide-β-D-glucuronide another prodrug is hydrolyzed by colonic bacterial and mucosal beta-glucuronidase to librate free budesonide into the colonic milieu. Hydrolysis rates of budesonide-β-D-glucuronide in fecal specimens from IBD patients and normal volunteers are similar. pH reduction in the colon of IBD patients may inhibit the bacterial hydrolysis of this prodrug.
Antioxidants
Antioxidant status has been described to be compromised in the intestinal mucosa from patients with IBD [46,47,86,87]. In vitro studies using enterocytes have also demonstrated the damaging effects of exposure to oxidants [88]. Similar studies reveal that the colons of IBD patients produce more oxygen free radicals compared with those of control subjects [57,48]. In addition, using a chemiluminescence assay, significantly elevated levels of reactive oxygen metabolites were found in the actively inflamed mucosa of IBD patients [47,89]. Colitis animals showed an increase in plasma-oxidized proteins, indicating that DSS induced oxidative stress to be at the systemic level that persisted until the end of the treatment [90,91,92,93,94]. Oxidative stress is linked with the stimulation of the immune system [18].
Glutathione (GSH)
The endogenous tripeptide glutathione (GSH), is the most important intracellular defense agent in mammalian organs, including the gut. GSH is involved in protein folding and protein synthesis, as well as in intracellular signaling [90]. Glutathione is the most abundant cellular antioxidant synthesized by animal cells. GSH plays an essential role in cell biology and modulates cell responses to redox changes associated with the presence of ROS. GSH neutralizes free radicals and ROS toxicity and is essential for both the functional and structural integrity of the gut. GSH may be depleted during inflammatory illness such as IBD [92,94] and acetaminophen toxicity [95,96,97] and this GSH deficiency predisposes animals to organ failure and death after an otherwise nonlethal period of hypotension [89,98]. Indeed GSH-deficient mice show severe degradation of the jejunum and colonic mucosa and have body weight loss and diarrhea [89]. Depletion of GSH induced enlarged lymphoid aggregates in the intestine by recruitment of lymphocytes from the peripheral circulation [99]. The depletion in tissue levels of GSH has been implicated as a component of the inflammatory complications. Supplementation with GSH monoethylester has been reported to prevent these lymphoid aggregates [100]. Data showed a significant decrease in the blood as well as intestinal tissue levels of GSH in the DSS-induced colitis model [94]. GSH plays an essential role in cell biology and modulates cell responses to redox changes associated with the presence of ROS. In vivo studies indicated that oxidized colonic mucosal redox status stimulated colonic mucosal growth in rats. The data supported that GSH is required to maintain normal colonic and plasma cysteine/cystine homeostasis [100]. Because GSH deficiency is associated with severe injury such as inflammation and sepsis, treatment strategies that maintain GSH levels may decrease the incidence of organ failure. One strategy for preventing this injury is to boost the levels of GSH within the cells. However, GSH is not taken up directly by most cell phenotypes and must first be broken down into its component amino acids (cysteine, glutamate, and glycine) by enzymes including δ-glutamyltranspeptidase.
Cysteine Prodrugs:
Cysteine an extracellular reducing agent, is a critical substrate for protein synthesis, and the rate-limiting precursor to GSH [101]. In vivo studies indicated that oxidized colonic mucosal redox status stimulated colonic mucosal growth in rats. Therefore, data suggested that GSH is required to maintain normal colonic and plasma cysteine /cystine homeostasis. Cysteine can be given orally to increase GSH [102] or to chelate trace elements in the gut, thereby decreasing absorption of both cysteine and the trace element [103].
Cysteine (CYSH) + Glutamate (GLU) = δ-glutamylcysteine (G-CLU-CYSH)
G-GLU-CYSH + Glycine (GLY) = GSH
2GSH + H2O2 = GSSG + 2H2O
GSSG + NADPH = 2GSH + NADP
Cysteine and glutathione prodrugs
Administration of N-acetylcysteine (NAC), a cysteine prodrug, in a rat model of chemically induced colitis (TNBS) attenuated acute colitis through increased mucosal GSH levels [104], suggesting that cysteine prodrugs may be beneficial in the acute relapse of IBD. Dextran sodium sulfate (DSS) treatment of mice mimics IBD in that it provokes inflammation and macrophage activation, with subsequent loss of epithelial integrity, and increases luminal Gram-negative microbiota [105]. Rebamipide, a drug that inhibits the production of free radicals, was shown to act as an anti-inflammatory agent in chronic DSS-induced colitis [106].
We investigated the efficacy of three different cysteine prodrugs against colitis in DDS-induced murine model. 2(RS)-n-Propylthiazolidine-4(R)-carboxylic acid (PTCA), is an L-cysteine prodrug that stimulate GSH biosynthesis with a masked sulfhydryl group in the form of a thiazolidine-4-carboxylic acid to stabilize it against oxidation (Figure 3a) [107]. PTCA undergoes ‘demand–pull’ hydrolysis and release GSH about 20% more efficiently than NAC [108]. 2(RS)-D-ribo-(1’,2’,3’,4’-tetrahydroxy- butyl)thiazolidine-4(R)-carboxylic acid, (RibCys) is another L-cysteine prodrug (D-Ribose-L-cysteine), that like PTCA, can liberate the sulfhydryl-containing amino acid L-cysteine in vivo by non-enzymatic, hydrolytic dissociation (Figure 3b) [100]. The ester of the carboxylic acid (carrier-linked prodrugs) is cleaved by hydrolysis (enzymatic and/or chemical) to liberate the active compound.
Figure 3.
Molecular structures of three cysteine and glutathione prodrugs: a) 2(RS)-n-Propylthiazolidine-4(R)-carboxylic acid (PTCA), an L-cysteine prodrug; b) 2(RS)-D-ribo-(1’,2’,3’,4’-tetrahydroxybutyl)thiazolidine-4(R)-carboxylic acid (RibCys) D-Ribose-L-cysteine and c) L-cysteine-glutathione mixed sulfide (CySSG).
Figure 3.
Molecular structures of three cysteine and glutathione prodrugs: a) 2(RS)-n-Propylthiazolidine-4(R)-carboxylic acid (PTCA), an L-cysteine prodrug; b) 2(RS)-D-ribo-(1’,2’,3’,4’-tetrahydroxybutyl)thiazolidine-4(R)-carboxylic acid (RibCys) D-Ribose-L-cysteine and c) L-cysteine-glutathione mixed sulfide (CySSG).

L-cysteine-glutathione mixed sulfide (CySSG) is a ubiquitous GSH derivative present in mammalian cells and has been suggested to be a depot form of GSH [109]. CySSG, a glutathione delivery agent [110] can liberate both GSH and L-cysteine by a glutathione reductase-catalyzed reaction with GSH (Figure 3c) [111].
We have shown that DSS-treated mice develop severe colitis, with decreased GSH concentrations in blood, colonic tissue and colon and liver when compared to normal controls [92,94,112]. In addition, DSS-treated animals had decreased hematocrits, and elevated inflammatory markers such as enzyme Cyclooxygenase (Cox-2), CD68, acute protein serum Amyloid A (SAA), and inflammatory cytokines (IL-6, IL-12, TNF-α) including osteopontin (OPN).
OPN is an important pro-inflammatory glycoprotein secreted by various cell types such as macrophages and intestinal epithelial cells [113]. In this study, PTCA-treated mice had significantly improved colonic lesions, with higher levels of blood and hepatic GSH, and lower GSSG concentrations compared to untreated DSS animals [114]. Based on our findings these antioxidant prodrugs reduced disease activity in the colitis. Oral administration of PTCA has been shown to protect against acetaminophen (APAP)-induced hepatic damage and necrosis, as well as GSH and cysteine depletion [92,115].
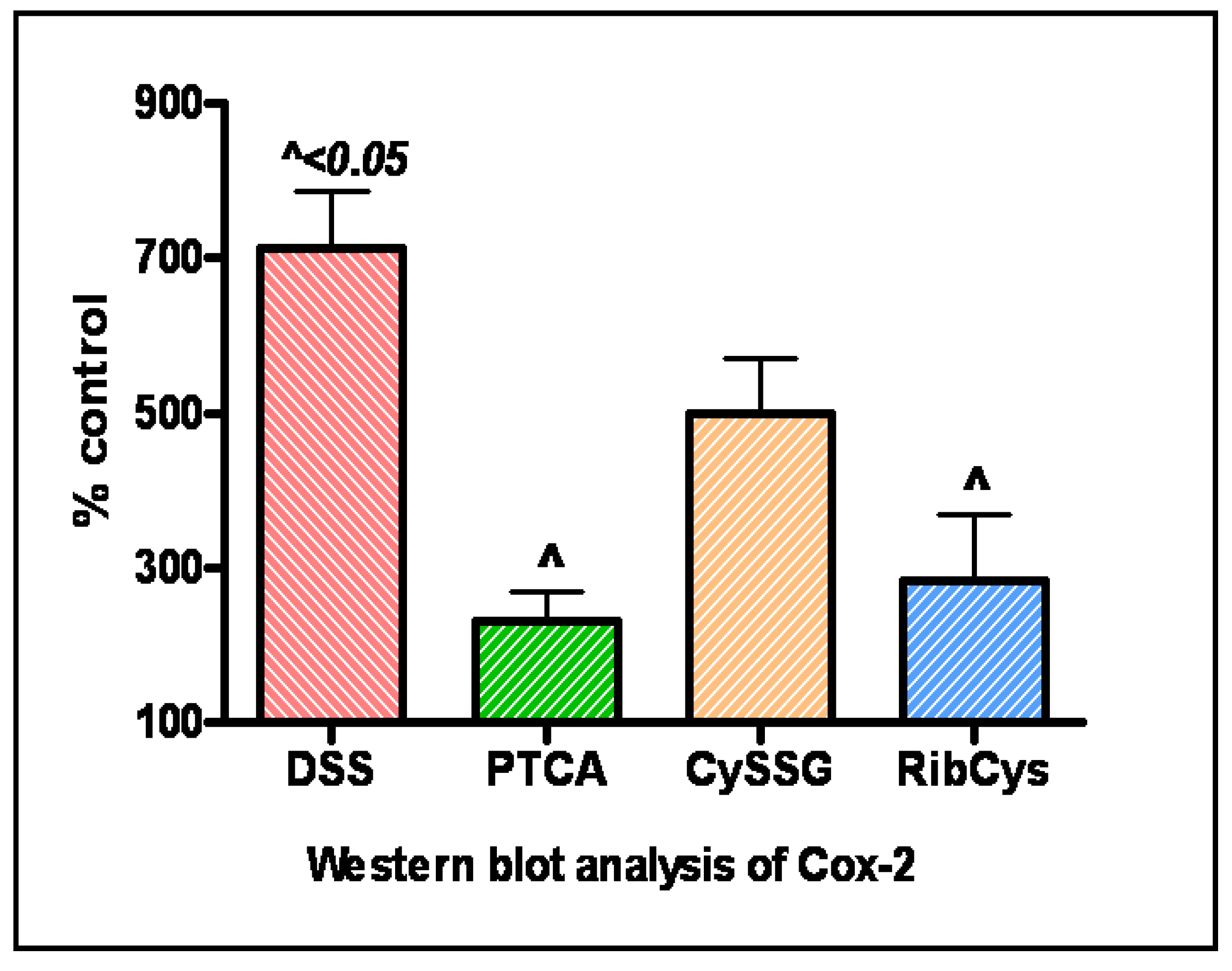
Figure 4.
Relative densities of representative western blot analyses (n= 3) indicating significant upregulation of Cyclooxygenase (Cox-2) protein expression in colonic tissue from colitis mice (DSS) which is normalized with prodrug therapy especially in PTCA animals. (DSS)- Represents colitis provided with DSS- supplemented water. PTCA, RibCys and CYSSG represent those mice provided with DSS- containing water and treated with one of cysteine/GSH prodrugs. (Courtesy of Oz et al, Translational Research 2007).
Figure 4.
Relative densities of representative western blot analyses (n= 3) indicating significant upregulation of Cyclooxygenase (Cox-2) protein expression in colonic tissue from colitis mice (DSS) which is normalized with prodrug therapy especially in PTCA animals. (DSS)- Represents colitis provided with DSS- supplemented water. PTCA, RibCys and CYSSG represent those mice provided with DSS- containing water and treated with one of cysteine/GSH prodrugs. (Courtesy of Oz et al, Translational Research 2007).

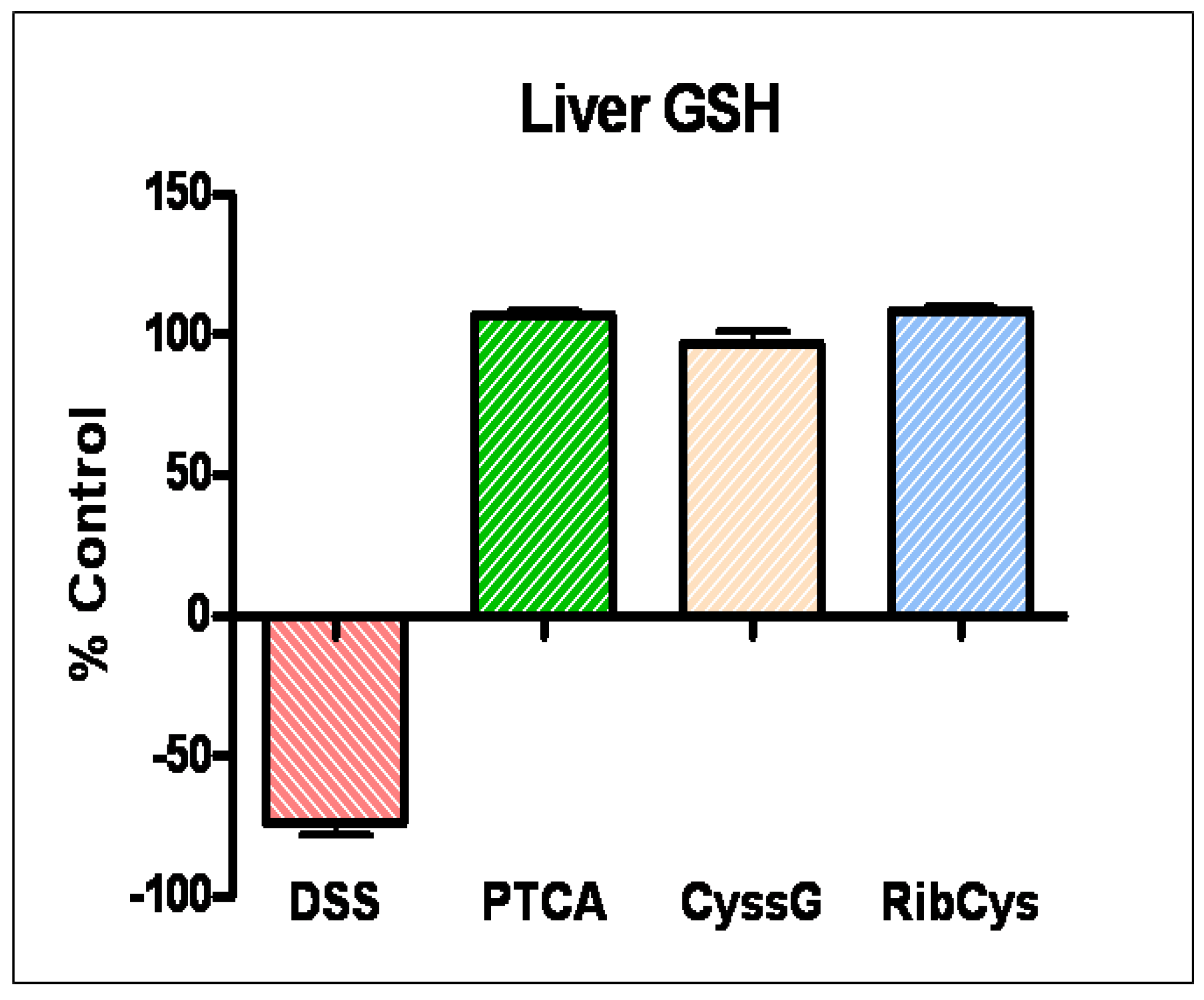
Figure 5.
Hepatic concentration of reduced GSH, the major source of gut antioxidant, was depleted in colitis animals (DSS) and normalized with prodrug therapy. (GSH was measured by HPLC).
Figure 5.
Hepatic concentration of reduced GSH, the major source of gut antioxidant, was depleted in colitis animals (DSS) and normalized with prodrug therapy. (GSH was measured by HPLC).

The results demonstrated a high degree of tissue selectivity in the APAP-induced depletion of GSH and cysteine concentrations, and in the effectiveness of PTCA in maintaining and even elevating sulfhydryl levels in extrahepatic tissues of APAP-treated mice [116], while the protective effect of PTCA was related to prevention of hepatic sulfhydryl depletion. Similarly, oral administration of PTCA provided protection against steatohepatitis by attenuating the expression of deleterious pro-inflammatory and fibrogenic genes in a dietary rat model [117]. Other studies showed that PTCA significantly increased GSH biosynthesis in cultured rat lens compared to untreated controls [118], and prevented naphthalene-induced experimental cataracts in mice [108]. Thus, our studies indicate that PTCA, RibCys and, to a lesser extent, CySSG, provide protection against colitis in this murine model. These data also reinforce our previous report describing the novel protective action of PTCA against different inflammatory complications including IBD [92,114,117].
S-adenosylmethionine (SAMe, Adomet) Metabolism and Gut
SAMe, an antioxidant and a GSH precursor is an obligatory intermediate in normal cell physiology and in the transsulfuration pathway. SAMe serves as an antioxidant not only because of its role as a precursor for GSH biosynthesis, but also because of its capacity to interact directly with ROS. The conversion of methionine to SAMe involves transfer of the adenosyl moiety of ATP to the sulfur atom of methionine [119]. This process is catalyzed by methionine adenosyltransferase (MAT) in cells [120]. The MAT gene is considered one of 33 genes required for survival and MAT is highly susceptible to oxidative stress [119]. Besides being a precursor for syntheses of polyamines, choline, and GSH, SAMe is also the major biologic methylating agent for DNA, RNA, phospholipids, biologic amines, and proteins. SAMe is required for the metabolism of nucleic acids, and for the maintenance of the structure and function of membranes [121]. SAMe in stable enteric capsule (cellulose gel) synthesized in the form of disulfate tosylate salt and, methacrylic acid copolymer has been shown to provide intestinal specific drug delivery systems with pH sensitive swelling and drug release properties. Methacrylic-type polymeric prodrugs have been shown to have a defined bioavailability.
Ulcerative colitis is associated with a selective reduction of n-butyrate oxidation by colonic epithelial cells [122,123]. The reasons for this are not completely understood. Bacterial production of anionic sulfide is reported to be increased in the colon of IBD patients and these sulfides can cause metabolic damage to colonocytes [123]. Incubation of colonic cell suspensions with sulfide induced a significant inhibition of 14CO2 production as compared with controls. Sulfide toxicity in isolated colonocytes can be reversed by methyl donors. The addition of S-adenosylmethionine-1,4-butane disulfonate to the cell suspension reversed this effect in proximal but not in distal cell incubations, suggesting a greater susceptibility of the distal colon to the sulfide effect [124].
The role of mucosal detoxification of sulfide by thiolmethyltransferase (TMT)-mediated methylation was suggested as crucial in protecting the healthy colonic mucosa from the adverse effects of luminal sulfide. In the human colon, methylation appears to protect colonic epithelial cells against sulfide-induced inhibition of n-butyrate oxidation. Luminal anionic sulfide may contribute to epithelial damage in ulcerative colitis [125]. Erythrocyte TMT activity was elevated in patients after proctocolectomy for Crohn's disease and Ulcerative Colitis. In patients with active stage of ulcerative colitis, the mucosal spermidine concentration was increased due to exogenous uptake in comparison with patients in remission or in healthy controls. The activities of ornithine decarboxylase and S-adenosylmethionine decarboxylase, rate-limiting enzymes of polyamine biosynthesis, were lower in the patients with ulcerative colitis [125]. These findings in patients suggest that sulfide detoxification may be important in the pathogenesis of IBD. Recent investigations show a close interaction between endogenous SAMe concentrations (measured by HPLC) and cytokine production in animal models [92,97,114,117,126]. SAMe deficiency increased TNFα production and tissue injury while exogenous SAMe supplementation decreased TNFα production in these animals. SAMe supplementation significantly improved blood level of GSH, and serum SAA level, as well as pathological scores in colitis [92] suggesting a therapeutic value in treatment of IBD. Oral administration of SAMe protected against acetaminophen hepatotoxicity in mice [97]. In addition, we reported a therapeutic effect of SAMe in oral form against liver injury, elevated liver enzymes aspartate transaminase and alanine transaminase and over expression of inflammatory and fibrotic genes (e.g. IL-1β, IL-6,TNFα, MMP3-9) in a dietary rat model for Nonalcoholic Steatohepatitis (NASH) [117,126]. We postulate that SAMe may also prove to be useful dietary supplement in the treatment of inflammatory disease in patients.
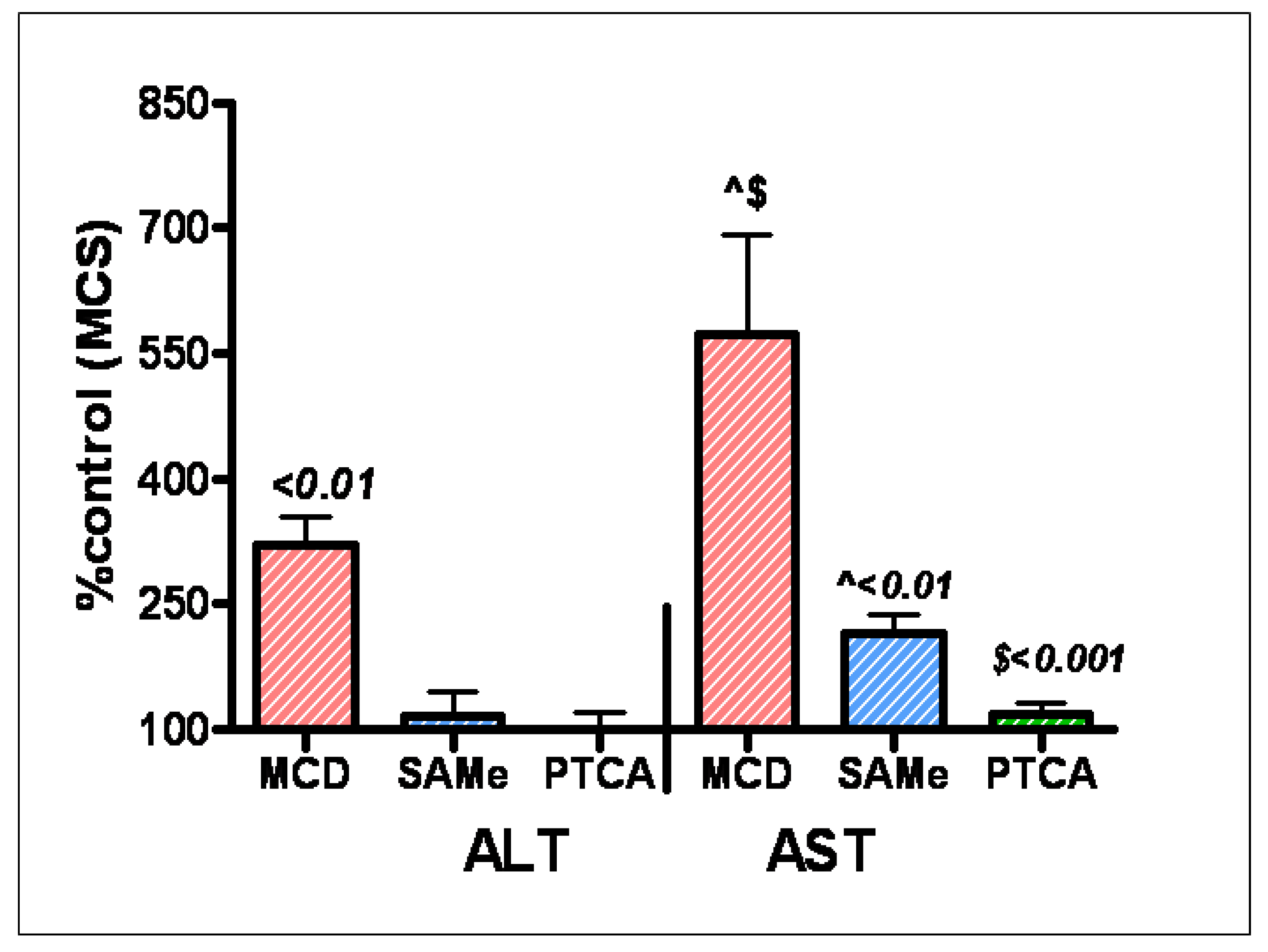
Figure 6.
Liver enzymes (ALT and AST) are significantly increased in rats on methionine Choline deficient diet (MCD) compared to those fed methionine choline sufficient diet (MCS). In contrast, SAMe and PTCA therapy normalize these deleterious enzyme activities in rats on an MCD diet (Model for steatohepatitis (NASH).
Figure 6.
Liver enzymes (ALT and AST) are significantly increased in rats on methionine Choline deficient diet (MCD) compared to those fed methionine choline sufficient diet (MCS). In contrast, SAMe and PTCA therapy normalize these deleterious enzyme activities in rats on an MCD diet (Model for steatohepatitis (NASH).

Conclusions
Oral delivery is the most common and preferred route of drug administration although the digestive tract exhibits several obstacles to drug delivery including motility and intraluminal pH profiles. Currently there is no cure for IBD. Although many treatments have been recommended for IBD, they do not treat the cause but are effective only in reducing the inflammation and accompanying symptoms [13,14,15,16,17,18,19,20,49,52,85]. Thus, current medical management of IBD consists of anti-inflammatory, immunosuppressive agents and other biologics. Since IBD patients undergo prolonged periods of treatment, prodrugs are considered as the most practical approach for targeting drugs to the colon in IBD patients. Three classes of prodrugs proven effective in IBD include: (1) anti-inflammatory agents (e.g. 5-ASA, azo prodrugs, amino acids) that can result from bacteria changing the function of prodrugs or prodrugs treating inflammation which changes the bacteria ecology; (2) immunomodulators and immunosuppressants; and (3) antioxidants. Improved drug delivery systems are required for drugs currently in use to treat localized diseases of the colon. Therefore, using IBD prodrug modalities to target specifically colonic tissue may lower the drug dosage, supply the drug only where required by maintaining the drug in its intact form close to the target site, and finally, reduce the incidence of systemic side effects in these patients.
Acknowledgements
The authors acknowledge NIH-NCCAM grant AT1490 and COBRE P20 RR020145 for financial support. The antioxidant prodrugs were synthesized by our collaborator Dr. Herbert Nagasawa at VA Medical Center, Minneapolis, Minn.
References
- Tursi, A.; Brandimarte, G.; Giorgetti, G.M.; Nasi, G. Assessment of orocaecal transit time in different localization of Crohn's disease and its possible influence on clinical response to therapy. Eur. J. Gastroenterol. Hepatol. 2003, 15, 69–74. [Google Scholar] [CrossRef]
- Schwab, M.; Klotz, U. Pharmacokinetic considerations in the treatment of inflammatory bowel disease. Clin. Pharmacokinet. 2001, 40, 723–751. [Google Scholar] [CrossRef]
- Dressman, J.B.; Vertzoni, M.; Goumas, K.; Reppas, C. Estimating drug solubility in the gastrointestinal tract. Adv. Drug Deliv. Rev. 2007, 59, 591–602. [Google Scholar] [CrossRef]
- Albert, A. Chemical aspects of selective toxicity. Nature 1958, 182, 421–422. [Google Scholar] [CrossRef]
- Stella, V.J.; Charman, W.N.A.; Naringrekar, V.H. Prodrugs. Do they have advantages in clinical practice? Drugs 1985, 29, 455–473. [Google Scholar] [CrossRef]
- Bundgaard, H. Design and application of prodrugs. In A Textbook of Drug Design and Development; Krogsgaard-Larsen, P., Bundgaard, H., Eds.; Harwood: Reading, UK, 1991; pp. 113–191. [Google Scholar]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004, 47, 2393–2404. [Google Scholar] [CrossRef]
- Han, H.K.; Amidon, G.L. Targeted prodrug design to optimize drug delivery. AAPS Pharm. Sci. 2000, 2, E6. [Google Scholar]
- Rubinstein, A. Approaches and opportunities in colon-specific drug delivery. Crit. Rev. Ther. Drug Carr. Syst. 1995, 12, 101–149. [Google Scholar] [CrossRef]
- Hovgaard, L.; Brondsted, H. Current applications of polysaccharides in colon targeting. Crit. Rev. Ther. Drug Carr. Syst. 1996, 13, 185–223. [Google Scholar] [CrossRef]
- Fang, L.; Battisti, R.F.; Cheng, H.; Reigan, P.; Xin, Y.; Shen, J; Ross, D.; Chan, K.K.; Martin, J.; Wang, P.G.; Sun, D. Enzyme specific activation of benzoquinone ansamycin prodrugs using HuCC49DeltaCH2-beta-Galactosidase conjugates. J. Med. Chem. 2006, 49, 6290–6297. [Google Scholar] [CrossRef]
- Curini, M.; Epifano, F.; Genovese, S. Synthesis of a novel prodrug of 3-(4′-geranyloxy-3′-methoxyphenyl)-2-trans-propenoic acid for colon delivery. Bioorg. Med. Chem. Lett. 2005, 15, 5049–5052. [Google Scholar] [CrossRef]
- Podolskiy, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef]
- Hutfless, S.M.; Weng, X.; Liu, L.; Allison, J.; Herrinton, L.J. Mortality by Medication use among patients with inflammatory bowel disease, 1996-2003. Gastroenterology 2007, 133, 1779–1786. [Google Scholar] [CrossRef]
- Cohen, R.D.; Woseth, D.M.; Thistel, R.A.; Hanauer, S.B. A meta-analysis and overview of the literature on treatment options for left-sided ulcerative colitis and ulcerative proctitis. Am. J. Gastroenterol. 2000, 95, 1263–1276. [Google Scholar] [CrossRef]
- Li, M.C.; He, S.H. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J. Gastroenterol. 2004, 10, 620–625. [Google Scholar]
- Neurath, M.F.; Pettersson, S.; Strober, W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kB abrogates established experimental colitis in mice. Nature Med. 1996, 2, 998–1004. [Google Scholar] [CrossRef]
- Neuman, M.G. Immune dysfunction in inflammatory bowel disease. Transl. Res. 2007, 149, 173–186. [Google Scholar] [CrossRef]
- Kozuch, P.L.; Hanauer, S.B. General principles and pharmacology of biologics in inflammatory bowel disease. Gastroenterol. Clin. North Am. 2006, 35, 757–773. [Google Scholar] [CrossRef]
- Kozuch, P.L.; Hanauer, S.B. Treatment of inflammatory bowel disease: A review of medical therapy. World J. Gastroenterol. 2008, 4, 354–377. [Google Scholar] [CrossRef]
- Sartor, R.B. Enteric microflora in IBD: pathogens or commensals? Inflamm. Bowel Dis. 1997, 3, 230–235. [Google Scholar] [CrossRef]
- Robertson, D.J; Sandler, R.S. Measles virus and Crohn’s disease: a critical appraisal of the current literature. Inflamm. Bowel Dis. 2001, 7, 51–57. [Google Scholar] [CrossRef]
- Sartor, RB. Microbial factors in the pathogenesis of Crohn’s disease, ulcerative colitis, and experimental intestinal inflammation. In InInflammatory Bowel Disease; Kirsner, J.B., Shorter, R.G., Eds.; Williams and Wilkens: Baltimore, 1995; pp. 96–124. [Google Scholar]
- Elson, C.O.; Sartor, R.B.; Tennyson, G.S.; Riddell, R.H. Insights into the pathogenesis of IBD provided by new rodent models of colitis. Inflam. Bowel Dis. 1995, 1, 64–75. [Google Scholar]
- Sadlack, B.; Merz, H.; Schorle, H.; Schimpl, A.; Feller, A.C.; Horak, I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993, 75, 255–261. [Google Scholar]
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in IL-10-deficient mice. Infect. Immun. 1998, 66, 5224–5231. [Google Scholar]
- MacDermott, R.P.; Stenson, W.F. Inflammatory Bowel Disease. In Immunology and Immunopathology of the liver and gastrointestinal tract; Targan, S.R., Shanahan, F., Eds.; Igaku-Shoin: New York, 1990; pp. 459–486. [Google Scholar]
- Gaetke, L.M.; Frederich, R.C.; Oz, H.S.; McClain, C.J. Zinc deficiency induced changes in plasma leptin, metabolic rate, and physical activity in rats. J. Nutr. Biochem. 2002, 13, 237–244. [Google Scholar] [CrossRef]
- Grave, G. Antioxidant nutrients in inflammatory bowel disease. In Progress in inflammatory bowel disease, Trends in IBD Care; CCFA: New York, 1994; pp. 12–14. [Google Scholar]
- Hatoum, O.A.; Binion, D.G.; Gutterman, D.D. Paradox of simultaneous intestinal ischaemia and hyperaemia in inflammatory bowel disease. Eur. J. Clin. Invest. 2005, 35, 599–609. [Google Scholar] [CrossRef]
- Berg, R.D. The indigenous gastrointestinal microflora. Trends Microbiol. 1996, 4, 430–435. [Google Scholar] [CrossRef]
- Shanahan, F. Probiotics in inflammatory bowel disease – therapeutic rationale and role. Drug Deliv. Rev. 2004, 56, 809–818. [Google Scholar] [CrossRef]
- Wang, J.H.; Redmond, H.P.; Watson, R.W.; Bouchei-Hayes, D. Role of lipopolysaccharide and tumor necrosis factor-alpha in induction of hepatocyte necrosis. Am. J. Physiol. 1995, 269, G297–G304. [Google Scholar]
- Beutler, B.; Kruys, V. Lipopolysaccharide signal transduction, regulation of tumor necrosis factor biosynthesis, and signaling by tumor necrosis factor itself. J. Cardiovasc. Pharmacol. 1995, 25 (suppl. 2), S1–S8. [Google Scholar] [CrossRef]
- Pastor Rojo, O.; López San Román, A.; Albéniz Arbizu, E.; de la Hera Martínez, A.; Ripoll Sevillano, E.; Albillos Martínez, A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 269–277. [Google Scholar] [CrossRef]
- Gaetke, L.; Oz, H.S.; Frederich, R.; McClain, C. Anti-TNF-α Antibody Normalizes Serum Leptin in IL-2 Deficient Mice. J. Amer. Coll. Nutri. 2003, 22, 415–420. [Google Scholar] [CrossRef]
- Conner, E.M.; Brand, S.; Davis, J.M.; Laroux, F.S.; Palombella, V.J.; Fuseler, J.W.; Kang, D.Y.; Wolf, R.E.; Grisham, M.B. Proteasome inhibition attenuates nitric oxide synthase expression, VCAM-1 transcription and the development of chronic colitis. J. Pharmacol. Exp. Ther. 1997, 282, 1615–1622. [Google Scholar]
- Scheinman, R.I.; Cogswell, P.C.; Lofquist, A.K.; Baldwin, A.S. Role of transcriptional activation of IkB-α in mediation of immunosuppression by glucocorticoids. Science 1995, 270, 283–286. [Google Scholar]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: inhibition of NF-ĸB activity through induction of IkB synthesis. Science 1995, 270, 286–290. [Google Scholar]
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J. Clin. Invest. 1998, 101, 1163–1174. [Google Scholar] [CrossRef]
- Yang, F.; de Villiers, W.J.S.; McClain, C.J.; Varilek, G.W. Mesalamine (5-ASA) modulates NF-kB/ IkB-α activity In Caco-2 cells. Gastroenterology 1997, 112, A1125. [Google Scholar]
- Meyer, S.; Kohler, N.G.; Joly, A. Cyclosporine A is an uncompetitive inhibitor of proteasome activity and prevents NF-κB activation. FEBS Lett. 1997, 413, 354–358. [Google Scholar] [CrossRef]
- Grisham, M.B.; Granger, D.N. Neutrophil-mediated mucosal injury. Role of reactive oxygen metabolites. Dig. Dis. Sci. 1988, 33, 6S–15S. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef] [Green Version]
- Babbs, C.F. Oxygen radicals in ulcerative colitis. Free Radic. Biol. Med. 1992, 13, 169–181. [Google Scholar] [CrossRef]
- Suematsu, M.; Suzuki, M.; Kitahora, T.; Miura, S.; Suzuki, K.; Hibi, T.; Watanabe, M.; Nagata, H.; Asakura, H.; Tsuchiya, M. Increased respiratory burst of leukocytes in inflammatory bowel diseases–the analysis of free radical generation by using chemiluminescence probe. J. Clin. Lab. Immunol. 1987, 24, 125–128. [Google Scholar]
- Simmonds, N.J.; Allen, R.E.; Stevens, T.R.; Van Someren, R.N.; Blake, D.R.; Rampton, D.S. Chemiluminescence assay of mucosal reactive oxygen metabolites in inflammatory bowel disease. Gastroenterology 1992, 103, 186–196. [Google Scholar]
- Shiratora, Y.; Aoki, S.; Takada, H.; Kiriyama, H.; Ohto, K.; Hai, K.; Teraoka, H.; Matano, S.; Matsumoto, K.; Kamii, K. Oxygen-derived free radical generating capacity of polymorphonuclear cells in patients with ulcerative colitis. Digestion 1989, 44, 163–171. [Google Scholar] [CrossRef]
- Stein, R.B.; Hanauer, S.B. Comparative tolerability of treatments for inflammatory bowel disease. Drug Saf. 2000, 23, 429–448. [Google Scholar] [CrossRef]
- Dubuquoy, L.; Rousseaux, C.; Thuru, X.; Peyrin-Biroulet, L.; Romano, O.; Chavatte, P.; Chamaillard, M.; Desreumaux, P. PPARγ as a new therapeutic target in inflammatory bowel disease. Gut 2006, 55, 1341–1349. [Google Scholar] [CrossRef]
- Sands, B.E. Therapy of inflammatory bowel disease. Gastroenterology 2000, 118, S68–S82. [Google Scholar] [CrossRef]
- Kinget, R.; Kalala, W.; Vervoort, L.; Mooter, G.U. Colonic drug targeting. J. Drug. Target. 1998, 6, 129–149. [Google Scholar] [CrossRef]
- Scheline, R. Metabolism of foreign compounds by gastrointestinal microorganisms. Pharmacol. Rev. 1973, 25, 451–523. [Google Scholar]
- Hanauer, S.B. Review article: high-dose aminosalicylates to induce and maintain remissions in ulcerative colitis. Aliment. Pharmacol. Ther. 2006, Suppl 3, 37–40. [Google Scholar] [CrossRef]
- Staerk Laursen, L.; Stokholm, M.; Bukhave, K.; Rask-Madsen, J.; Lauritsen, K. Disposition of 5-aminosalicylic acid by olsalazine and three mesalazine preparations in patients with ulcerative colitis: comparison of intraluminal colonic concentrations, serum values, and urinary excretion. Gut 1990, 31, 1271–1276. [Google Scholar] [CrossRef]
- Dhaneshwar, S.S.; Gairola, N.; Kandpal, M.; Vadnerkar, G.; Bhatt, L. Colon-specific, mutual azo prodrug of 5-aminosalicylic acid with L-tryptophan: synthesis, kinetic studies and evaluation of its mitigating effect in trinitrobenzenesulfonic acid- induced colitis in rats. Bioorg. Med. Chem. 2007, 15, 4903–4909. [Google Scholar] [CrossRef]
- Bishop, J.; Furman, M.; Thomson, M. Omeprazole for gastroesophageal reflux disease in the first 2 years of life: a dose-finding study with dual-channel pH monitoring. J. Pediatr. Gastroenterol. Nutr. 2007, 45, 50–55. [Google Scholar] [CrossRef]
- Oosterhuis, B.; Jonkman, J.H. Omeprazole: pharmacology, pharmacokinetics and interactions. Digestion 1989, 44 suppl, 9–17. [Google Scholar] [CrossRef]
- Topping, D.L.; Clifton, P.M. Short chain fatty acids and human colonic function: roles of resistant starch and non-starch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar]
- Brouns, F.; Kettlitz, B.; Arrigoni, E. Resistant starch and "butyrate revolution". Trends Food Sci. Technol. 2002, 13, 251–261. [Google Scholar] [CrossRef]
- Caderni, G.; Luceri, C.; Lancioni, L.; Tessitore, L.; Dolara, P. Slow-release pellets of sodium butyrate increase apoptosis in the colon of rats treated with azoxymethane, without affecting aberrant crypt foci and colonic proliferation. Nutr. Cancer 1998, 30, 175–181. [Google Scholar] [CrossRef]
- Scheppach, W.; Sommer, H.; Kirchner, T.; Paganelli, G.M.; Bartram, P.; Christl, S.; Richter, F.; Dusel, G.; Kasper, H. Effect of butyrate enema on the colonic mucosa in distal ulcerative colitis. Gasteroentrology 1992, 103, 51–56. [Google Scholar]
- Illman, R.J.; Topping, D.L.; McIntosh, G.H.; Trimble, R.P.; Storer, G.B.; Taylor, M.N. The hypocholesterolaemic effects of dietary propionate: studies in whole animals and perfused rat liver. Ann. Nutr. Metab. 1988, 32, 97–107. [Google Scholar] [CrossRef]
- Annison, G.; Illman, R.J.; Topping, D.L. Acetylated propionylated or butyrylated starches raise large bowel short-chain fatty acids preferentially when fed to rats. Nutrition 2003, 133, 3523–3528. [Google Scholar]
- Bennett, M.J.; Rinaldo, P.; Strauss, A.W. Inborn errors of mitochondrial fatty acid oxidation. Crit. Rev. Clin. Lab. Sci. 2000, 37, 1–44. [Google Scholar] [CrossRef]
- Rinaldo, P. Fatty acid transport and mitochondrial oxidation disorders. Semin. Liver Dis. 2001, 21, 489–500. [Google Scholar] [CrossRef]
- Rinaldo, P.; Matern, D.; Bennett, M.J. Fatty acid oxidation disorders. Annu. Rev. Physiol. 2002, 64, 477–502. [Google Scholar] [CrossRef]
- Srinivas, S.R.; Prasad, P.D.; Umapathy, N.D.; Ganapathy, V.; Shekhawat, P.S. Transport of butyryl-L-carnitine, a potential prodrug, via the carnitine transporter OCTN2 and the amino acid transporter ATB0,+. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1046–G1053. [Google Scholar]
- Lipsky, J.J. Mycophenolate mofetil. Lancet 1996, 348, 1357–1359. [Google Scholar] [CrossRef]
- Gosio, B. Riceche bacteriologiche e chimiche sulle alterazioni del mais. Riv. Igiene. Sanita Pubblica 1896, 7, 825–868. [Google Scholar]
- Hupe, D.J.; Azzolina, B.A.; Behrens, N.D. IMP dehydrogenase from the intracellular parasitic protozoan Eimeria Tenella and its inhibition by mycophenolic acid. J. Biol. Chem. 1986, 261, 8363–8369. [Google Scholar]
- Oz, H.S.; Hughes, W. Novel anti-Pneumocystis carinii effects of the immunosuppressant mycophenolate mofetil in contrast to provocative effects of tacrolimus, sirolimus and dexamethasone. J. Infect. Dis. 1997, 175, 901–904. [Google Scholar] [CrossRef]
- Oz, H.S.; Hughes, W.T.; Varilek, G. Provocative effects of immunosuppressants rapamycin, tacrolimus and dexamethasone on P. carinii pneumonitis in contrast to anti-PCP effects of the mycophenolate mofetil. Transplant 2001, 72, 1464–1465. [Google Scholar] [CrossRef]
- Azevedo, L.S.; Castro, M.C.R.; Paula, F.J.; Ianhez, L.E.; David-Neto, E. Mycophenolate mofetil may protect against Pneumocystis carinii pneumonia in renal patients. Rev. Inst. Med. Trop. S. Paulo 2005, 47, 143–145. [Google Scholar] [CrossRef]
- Zeeh, J.M.; Zorlu, Riley, N.E.; Hoffmann, P.; Ruwe, M.; Goebell, H.; Gerken, G.; Dignass, A.U. Mycophenolate mofetil reduces tissue damage and inflammation in an experimental model of colitis in rats. Scand. J. Gastroenterol. 2001, 36, 66–70. [Google Scholar]
- Neurath, M.F.; Wanitschke, R.; Peters, M.; Krummenauer, F.; Meyer, zum Buschenfelde, K.H.; Schlaak, J.F. Randomised trial of mycophenolate mofetil versus azathioprine for treatment of chronic active Crohn’s disease [comment]. Gut 1999, 44, 625–628. [Google Scholar] [CrossRef]
- Orth, T.; Peters, M.; Schlaak, J.F.; Krummenauer, F.; Wanitschke, R.; Mayet, W.J.; Galle, P.R.; Neurath, M.F. Mycophenolate mofetil versus azathioprine in patients with chronic active ulcerative colitis: a 12-month pilot study. Am. J. Gastroenterol. 2000, 95, 1201–1207. [Google Scholar] [CrossRef]
- Florin, T.H.; Roberts, R.K.; Watson, M.R.; Radford-Smith, G.L. Treatment of steroid refractory inflammatory bowel disease (IBD) with mycophenolate mofetil (MMF). Aust. N. Z. J. Med. 1998, 28, 344–345. [Google Scholar] [CrossRef]
- Miehsler, W.; Reinisch, W.; Moser, G.; Gangl, A.; Vogelsang, H. Is mycophenolate mofetil an effective alternative in azathioprine-intolerant patients with chronic active Crohn’s disease. Am. J. Gastroenterol. 2001, 96, 782–787. [Google Scholar] [CrossRef]
- Golconda, M.S.; Valente, J.F.; Bejarano, P.; Gilinsky, N.; First, MR. Mycophenolate mofetil induced colonic ulceration in renal transplant recipients. Transplant. Proc. 1999, 31, 272–273. [Google Scholar] [CrossRef]
- Shaw, L.M.; Nowak, I. Mycophenolic acid: measurement and relationship to pharmacologic effects. Ther. Drug Monit. 1995, 17, 685–689. [Google Scholar] [CrossRef]
- Loftus, C.G.; Egan, L.J.; Sandborn, W.J. Cyclosporine, tacrolimus, and mycophenolate mofetil in the treatment of inflammatory bowel disease. Gastroenterol. Clin. N. Am. 2004, 33, 141–169. [Google Scholar] [CrossRef]
- Breen, D.P.; Marinaki, A.M.; Arenas, M.; Hayes, P.C. Pharmacogenetic association with adverse drug reactions to azathioprine immunosuppressive therapy following liver transplantation. Liver Transpl. 2005, 11, 826–833. [Google Scholar] [CrossRef]
- Marín-Jimenez, I.; Pena, A.S. Budesonide for ulcerative colitis. Rev. Esp. Enferm. Dig. 2006, 98, 362–373. [Google Scholar]
- Parks, D.A.; Bulkley, G.B.; Granger, D.N. Role of oxygen-derived free radicals in digestive tract diseases. Surgery 1983, 94, 415–422. [Google Scholar]
- Tsuchiya., M. Free radicals in digestive diseases. In Proceedings of the 1st International Symposium on Free Radicals in Digestive Diseases; Excerpta Medica: New York, 1988; p. 244. [Google Scholar]
- Baker, S.S.; Campbell, C.L. Enterocyte injury by O2-dependent processes. Gastroenterology 1991, 101, 716–720. [Google Scholar]
- Mrtensson, J.; Jain, A.; Meister, A. Glutathione is required for intestinal function. Proc. Nat. Acad. Sci. 1990, 87, 1715–1719. [Google Scholar] [CrossRef] [Green Version]
- Uhlig, S.; Wendel, A. The physiological consequences of glutathione variations. Life. Sci. 1992, 51, 1083–1094. [Google Scholar] [CrossRef]
- Mercier, S.; Breuille, D.; Mosoni, L.; Obled, C.; Patureau Mirand, P. Chronic Inflammation Alters Protein Metabolism in Several Organs of Adult Rats. J. Nutr. 2002, 132, 1921–1928. [Google Scholar]
- Oz, H.S.; Chen, T.; McClain, C.J.; deVilliers, W. Antioxidants a novel therapy in a murine model of colitis. J. Nutri. Biochem. 2005, 16, 297–304. [Google Scholar] [CrossRef]
- Dieleman, L.A.; Palmen, M. J.; Akol, H.; Bloemena, E.; Pena, A.S.; Meuwissen, S.G.; et al. Chronic experimental colitis induced by dextran sulfate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 1998, 114, 385–391. [Google Scholar] [CrossRef]
- Oz, H.S.; Chen, T.; de Villiers, W.; McClain, C. Metallothionein overexpression does not protect against inflammatory bowel disease in a DSS murine colitis model. Med. Sci. Monit. 2005, 11, BR69–73. [Google Scholar]
- Sido, B.; Hack, V.; Hochlehnert, A.; Lipps, H.; Herfarth, C.; Dröge, W. Impairment of intestinal glutathione synthesis in patients with inflammatory bowel disease. Gut 1998, 42, 485–492. [Google Scholar] [CrossRef]
- Chen, T.S.; Richie, J.P., Jr; Lang, C.A. Life span profiles of glutathione and acetaminophen detoxification. Drug Metab. Disp. 1990, 18, 882–887. [Google Scholar]
- Oz, H.S.; McClain, C.; Nagasawa, H.; Ray, M.; Chen, T. Diverse Antioxidants protect Against Acetaminophen Hepatotoxicity. J. Biochem. Mol. Tox. 2004, 18, 361–368. [Google Scholar]
- Robinson, M.K.; Rounds, J.D; Hong, R.W.; Jacobs, D.O.; Wilmore, D.W. Glutathione deficiency increases organ dysfunction after hemorrhagic shock. Surgery 1992, 112, 140–7, 148–149. [Google Scholar]
- Koch, T.R.; Yuan, L.X.; Fink, J.G.; Petro, A.; Opara, E.C. Induction of enlarged intestinal lymphoid aggregates during acute glutathione depletion in a murine model. Dig. Dis. Sci. 2000, 45, 2115–2121. [Google Scholar] [CrossRef]
- Tian, J.; Washizawa, N.; Gu, L.H.; Levin, M.S.; Wang, L.; Rubin, D.C.; Mwangi, S.; Srinivasan, S.; Gao, Y.; Jones, D.P.; Ziegler, T.R. Stimulation of colonic mucosal growth associated with oxidized redox status in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1081–R1091. [Google Scholar]
- Roberts, J.C.; Nagasawa, H.T.; Zera, R.T.; Fricke, R.F.; Goon, D.J. Prodrugs of L-cysteine as protective agents against acetaminophen-induced hepatotoxicity. 2-(Polyhydroxyalkyl)- and 2-(polyacetoxyalkyl)-thiazolidine-4(R)-carboxylic acids. J. Med. Chem. 1987, 30, 1891–1896. [Google Scholar] [CrossRef]
- Kleinman, W.A.; Richie Jr, J.P. Status of glutathione and other thiols and disulfides in human plasma. Biochem. Pharmacol. 2000, 60, 19–29. [Google Scholar] [CrossRef]
- Baker, D.H.; Czarnecki-Maulden, G.L. Pharmacologic role of cysteine in ameliorating or exacerbating mineral toxicities. J. Nutr. 1987, 117, 1003–1010. [Google Scholar]
- Ardite, E.; Sans, M.; Panes, J.; Romero, F.; Pique, J.M.; Fernandez-Checa, J. Replenishment of glutathione levels improves mucosal function in experimental acute colitis. Lab. Invest. 2000, 80, 735–744. [Google Scholar] [CrossRef]
- Kim, H.S.; Berstad, A. Experimental colitis in animal model. Scand. J. Gastroentrol. 1992, 27, 529–537. [Google Scholar] [CrossRef]
- Iwai, A.; Iwashita, E. Changes in colonic inflammation induced by dextran sulfate sodium (DSS) during short- and long-term administration of rebamipide. Dig. Dis. Sci. 1998, 43, 143S–147S. [Google Scholar]
- Nagasawa, H.T.; Goon, D.J.; Muldoon, W.P.; Zera, R.T. 2-Substituted thiazolidine-4(R)-carboxylic acids as prodrugs of L-cysteine, protection of mice against acetaminophen hepatotoxicity. J. Med. Chem. 1984, 27, 591–596. [Google Scholar] [CrossRef]
- Rathbun, W.B.; Nagasawa, H.T.; Killen, C.E. Prevention of naphthalene-induced cataract and hepatic glutathione loss by the L-cysteine prodrugs, MTCA and PTCA. Exp. Eye Res. 1996, 62, 433–441. [Google Scholar] [CrossRef]
- Butler, J.D.B.; Spielberg, S.P. Accumulation of cystine from glutathione-cysteine mixed disulfide in cystinotic fibroblasts; blockade by an inhibitor of γ-glutamyl transpeptidase. Life Sci. 1982, 31, 2563–2570. [Google Scholar] [CrossRef]
- Berkeley, L.I.; Cohen, J.F.; Crankshaw, D.L.; Shirota, F.; Nagasawa, H.T. Hepatoprotection by L-cysteine-glutathione mixed disulfide, a sulfhydryl-modified prodrug of glutathione. J. Biochem. Mol. Toxicol. 2003, 17, 95–97. [Google Scholar] [CrossRef]
- Eriksson, S.A.; Mannervik, B. The reduction of the L-cysteine-glutathione mixed disulfide in rat liver. Involvement of an enzyme catalyzing thiol-disulfide interchange. FEBS Lett. 1970, 7, 26–28. [Google Scholar] [CrossRef]
- Oz, H.S.; Ray, M.; Chen, T.S.; McClain, C.J. Efficacy of a TGF-β2 Containing Nutritional Support Formula in a Murine Model of IBD. J. Amer. Coll. Nutri. 2004, 23, 220–226. [Google Scholar] [CrossRef]
- Zhong, J.; Eckhardt, E.R.; Oz, H.S.; Bruemmer, D.; de Villiers, W.J. Osteopontin-deficiency protects mice from dextran sodium sulfate-induced colitis. Inflam. Bowel. Dis. 2006, 12, 1–7. [Google Scholar]
- Oz, H.S.; Chen, T.; Nagasawa, H. Comparative Efficacies of Two Cysteine Prodrugs and a Glutathione Delivery Agent in a Colitis Model. Translat. Res. 2007, 150, 122–129. [Google Scholar]
- Srinivasan, C.; Williams, W.M.; Ray, M.B.; Chen, T.S. Prevention of acetaminophen-induced liver toxicity by 2(R,S)-n-propylthiazolidine-4(R)-carboxylic acid in mice. Biochem. Pharmacol. 2001, 61, 245–252. [Google Scholar] [CrossRef]
- Srinivasan, C.; Williams, W.M.; Nagasawa, H.T.; Chen, T.S. Effects of 2(RS)-n-propylthiazolidine-4(R)-carboxylic acid on extrahepatic sulfhydryl levels in mice treated with acetaminophen. Biochem. Pharmacol. 2001, 61, 925–931. [Google Scholar] [CrossRef]
- Oz, H.S.; Im, H.J.; Chen, T.S.; de Villiers, W.J.; McClain, C.J. Glutathione enhancing agents protect against Steatohepatitis in a model. J. Biochem. Mol. Tox. 2006, 20, 39–47. [Google Scholar]
- Holleschau, A.M.; Rathbun, W.B.; Nagasawa, H.T. An HPLC radiotracer method for assessing the ability of L-Cysteine prodrugs to maintain glutathione levels in the cultured rat lens. Curr. Eye Res. 1996, 15, 510. [Google Scholar]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell. Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Giulidori, P.; Galli-Kienle, M.; Catto, E.; Stramentinoli, E. Transmethylation, transsulfuration, and aminopropylation reactions of S-adenosyl-L-methionine in vivo. J. Biol. Chem. 1984, 259, 4205–4211. [Google Scholar]
- Roediger, W.E.; Babidge, W.; Millard, S. Methionine derivatives diminish sulphide damage to colonocytes--implications for ulcerative colitis. Gut 1996, 39, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Roediger, W.E.; Babidge, W.J. Thiol methyltransferase activity in inflammatory bowel disease. Gut 2000, 47, 206–210. [Google Scholar] [CrossRef]
- Moore, J.W.; Babidge, W.J.; Millard, S.H.; Roediger, W.E. Thiolmethyltransferase activity in the human colonic mucosa: implications for ulcerative colitis. J. Gastroenterol. Hepatol. 1997, 12, 678–684. [Google Scholar] [CrossRef]
- Obayashi, M.; Matsui-Yuasa, I.; Matsumoto, T.; Kitano, A.; Kobayashi, K.; Otani, S. Polyamine metabolism in colonic mucosa from patients with ulcerative colitis. Am. J. Gastroenterol. 1992, 87, 736–740. [Google Scholar]
- Oz, H.S.; Chen, T.; Neuman, M. Methionine deficiency and liver injury in a dietary NASH model. Dig. Dis. Sci. 2007. [Google Scholar] [CrossRef] [Green Version]
- Sample Availability: Contact the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Oz, H.S.; Ebersole, J.L. Application of Prodrugs to Inflammatory Diseases of the Gut. Molecules 2008, 13, 452-474. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13020452
AMA Style
Oz HS, Ebersole JL. Application of Prodrugs to Inflammatory Diseases of the Gut. Molecules. 2008; 13(2):452-474. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13020452
Chicago/Turabian StyleOz, Helieh S., and Jeffrey L. Ebersole. 2008. "Application of Prodrugs to Inflammatory Diseases of the Gut" Molecules 13, no. 2: 452-474. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13020452