2. Results and Discussion
Starting from an open-chain alkene
2 and a halogenated arene
1 three reaction steps lead to the stereoselective formation of a styrene derivative in the presence of a stabilizing ligand L. The first step is a
syn-carbopalladation of the alkene giving
4, followed by rotation around the central single bond in
5 to allow the final step, the
syn-dehydropalladation giving a styrene derivative
3. In the presence of chiral ligands L
2*, such as sugar-based, chiral phosphite-oxazolines the Heck reaction of monocyclic alkenes like cyclopentene (
6) proceeds with high regio- and enantioselectivity generating one asymmetric center [
17] (
Scheme 1).
Scheme 1.
The Heck reaction of acyclic and monocyclic alkenes 2 and 6.
Scheme 1.
The Heck reaction of acyclic and monocyclic alkenes 2 and 6.
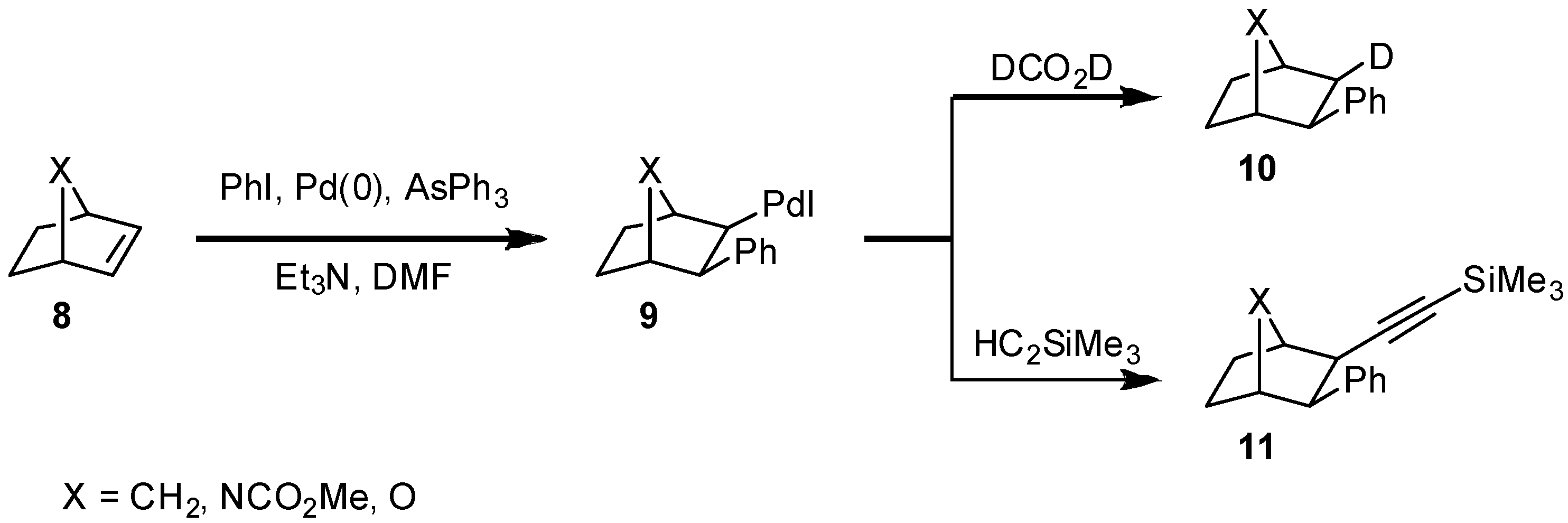
Rigid bicyclic alkenes
8 cannot be arylated this way as the final
syn-dehydropalladation of intermediate
9 is not feasible due to geometrical constraints; inter- or intramolecular consecutive reactions of the carbopalladation product
9 such as reduction or alkynylation (
10,
11) work well [
7]. Many applications for the reductive variant of the Heck reaction are known and the reductive arylation of bicyclic alkenes such as norbornene and its aza- or oxa-analogues
8 using palladium catalysts has been well studied [
18] (
Scheme 2).
Scheme 2.
Intermolecular consecutive reactions of a carbopalladation product 9 from 8.
Scheme 2.
Intermolecular consecutive reactions of a carbopalladation product 9 from 8.
Both, the carbopalladation to yield
9 and the consecutive substitution reactions proceed with complete
exo-diastereoselectivity. This was a very important finding for the stereoselective synthesis of the highly bioactive alkaloid epibatidine [
19], the parent amine of
13. Epibatidine, which is a lead in the development of new analgesics, is accessible by hydroarylation of the 7-azabicyclo-[2.2.1]heptene system with 6-chloro-3-iodopyridine. In the presence of chiral ligands, three asymmetric centers can be controlled. The epibatidine enantiomers show different biological activities. For biological testing, analogues modified in the 7-position such as
12,
14 have been synthesized. The enantiomeric excesses achieved are strongly dependent on the ligands employed. The P,P-ligand (
R)-BINAP (
15) is well suited for the synthesis of the epibatidine derivative
13 (81% e.e.), whereas the P,N-ligand (
S)-Ms-Valphos
16 proved to be the best ligand for the hydrohetarylation of the carbon analogue norbornene. Both ligands give low enantiomeric excesses in case of the oxa analogue, though (
Table 1).
Table 1.
Asymmetric hydro(hetero)arylation of bicyclo[2.2.1]heptene and its 7-hetero- analogues: e.e. (c.y.) [%] of the products 12-14.
Table 1.
Asymmetric hydro(hetero)arylation of bicyclo[2.2.1]heptene and its 7-hetero- analogues: e.e. (c.y.) [%] of the products 12-14.
![Molecules 15 03402 i001]() |
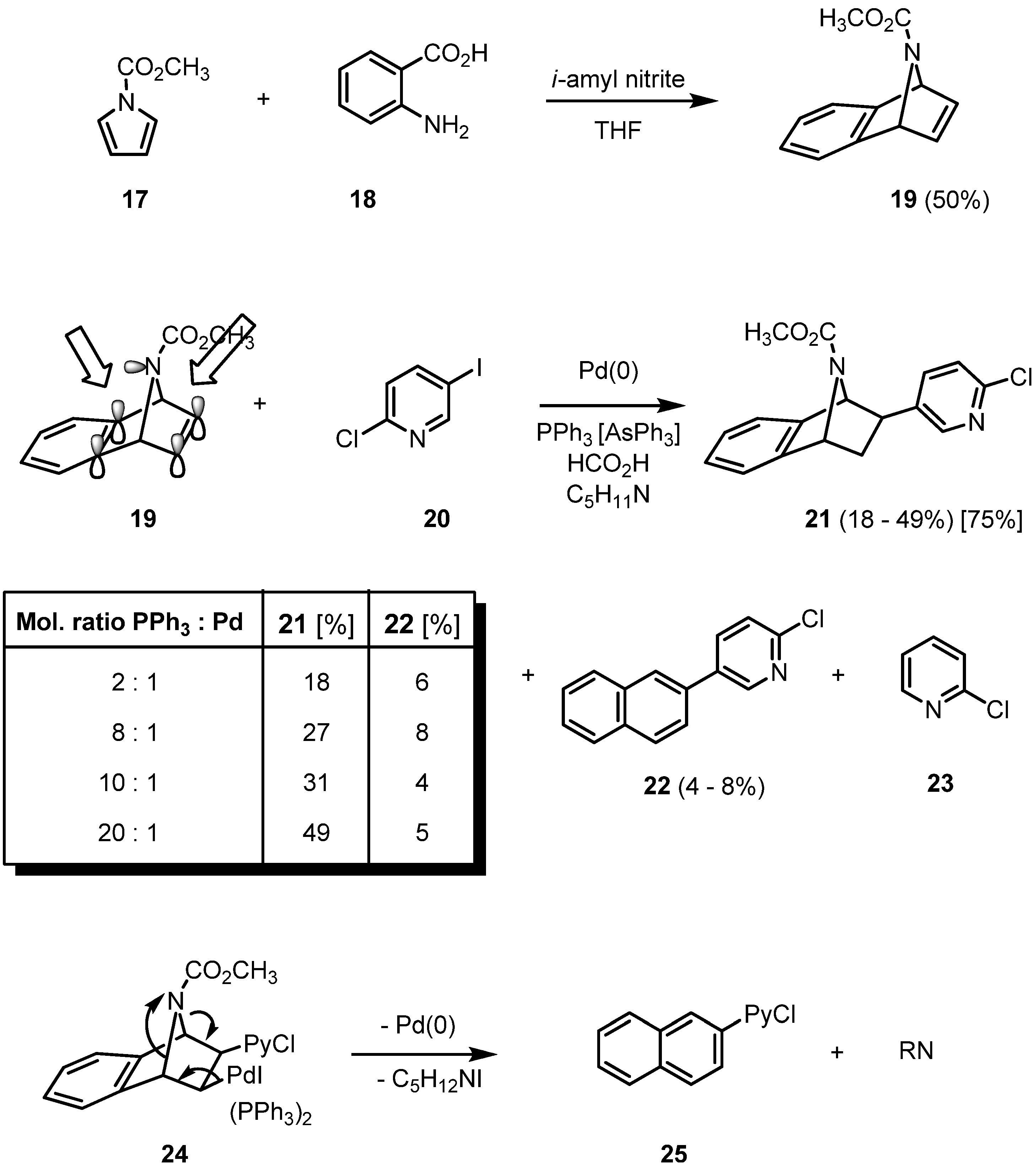
The reductive Heck arylation can be extended to the more lipophilic benzoannelated epibatidine analogues. They are easily accessible by a Diels-Alder reaction of cyclopentadiene, furan or
N-methoxycarbonylpyrrole (
17) with benzyne, generated from the diazotation product of anthranilic acid (
18). Using triphenylphosphine as a ligand, the mechanism of the hydroarylation of
19 becomes more complex (
Scheme 3).
Scheme 3.
Reductive Heck arylation of 19 with 2-chloro-5-iodopyridine (IPyCl, 20).
Scheme 3.
Reductive Heck arylation of 19 with 2-chloro-5-iodopyridine (IPyCl, 20).
When triphenylphosphine is employed as a ligand in Heck reactions, a two-fold phosphine-coordinated palladium complex is known to be the catalytically active species. In the case of the hydroarylation of
19, though, the yield of product
21 increases from 18 to 49% in parallel to an increase of the ligand to palladium ratio from 2:1 to 20:1 [
20]. This surprising result can only be rationalized by proposing a rather stable palladium complex between an aromatic π-bond and the nitrogen lone pair. NMR spectroscopic investigations indicate that the nitrogen inversion is already fast at room temperature. A higher amount of phosphine ligand increases the ligand pressure on the palladium, thus destabilizing the proposed complex formation on the “wrong” inner double bond of the bicycle and giving the active palladium species a fair chance to approach the free olefinic side of the azatricyclic system. Two side products are formed, a formal nitrene-extrusion product
22 of the initial carbopalladation product of the pyridylpalladium iodide to
19 and the product of a selective reduction of the chloroiodopyridine
20 giving
23. Additionally, the yield of
21 is strongly dependent on the nucleophilicity of the nitrogen: apparently, only electron-withdrawing groups such as the ester function of
19 prevent strong complexation and concomitant deactivitation of the palladium. While the parent system still gives the hydroarylation product in 28% yield, with an
N-methyl-substituent the corresponding coupling product cannot be isolated even in traces. The argument of competitive
N-complexation can also be applied when the unsuccessful reductive coupling of the pyridino-substituted analogue of
19 is discussed. The better stabilizing ligand triphenylarsine leads to the best yield of
21 (75%) [
18,
20].
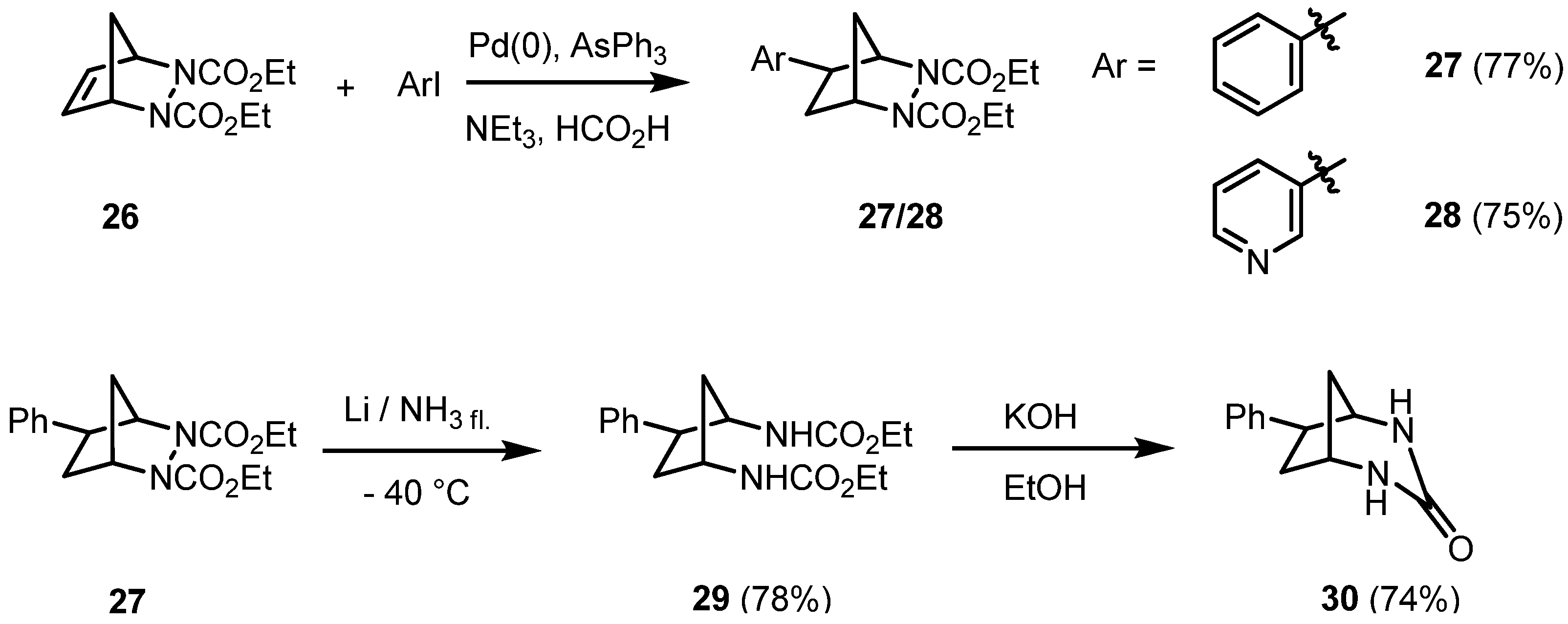
At that point we became interested in the scope and limitations of reductive Heck reactions of (bishetero)bicyclic alkenes. 2,3-Diazabicyclic alkenes are of special interest as the N-N and C-N bond represent an internal point of cleavage. The diazabicyclic alkene
26 is easily accessible by a Diels-Alder reaction of cyclopentadiene with diethyl azodicarboxylate (DEAD). Its reductive C-C coupling reaction with (het)aryl halides in the presence of an
in situ generated palladium catalyst, stabilized by triphenylarsine, afforded exclusively the
exo-hydroarylation products
27 and
28 in good yields [
21] (
Scheme 4).
Scheme 4.
Hydroarylation of the 2,3-diazabicyclo[2.2.1]heptene 26: stereoselective synthesis of intermediates.
Scheme 4.
Hydroarylation of the 2,3-diazabicyclo[2.2.1]heptene 26: stereoselective synthesis of intermediates.
Again, it is feasible to substitute the formate in a hydroorganylation reaction by the nucleophilic phenylacetylene as shown in
Scheme 2, resulting in a sequential C-C coupling reaction of
26. The reductive cleavage of the N-N bond yielded the
cis-1,3-diaminocyclopentane derivative
29 with an aryl substituent being
trans-oriented. Treatment of the bisurethane
29 with a base afforded the cyclic urea
30 in a good yield, as shown in
Scheme 4.
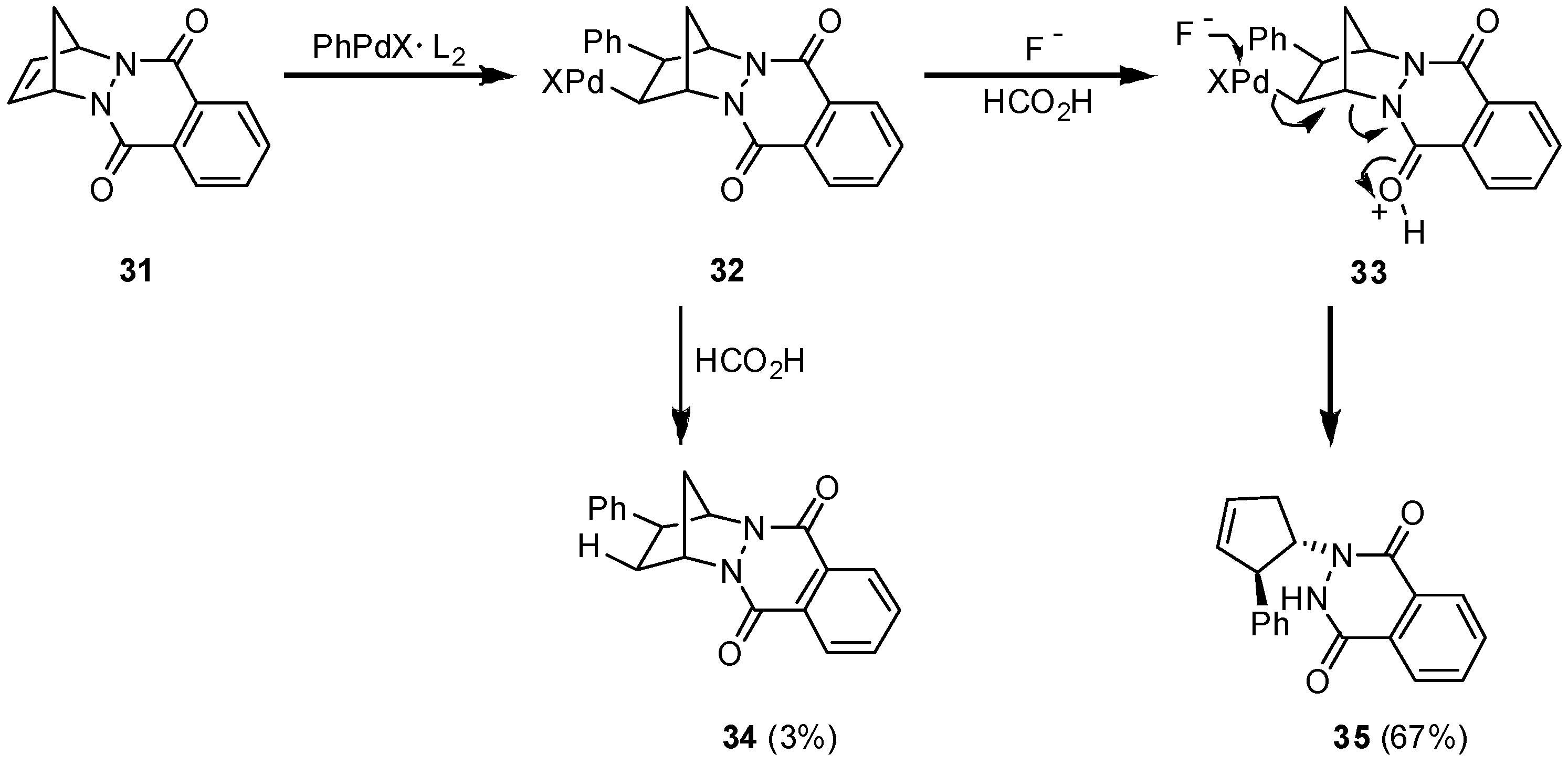
Encouraged by this result we tried the hydroarylation of the sterically more encumbered and more rigid tetracyclic Diels-Alder adduct
31 of 1,3-cyclopentadiene with the highly reactive azodienophile 1,4-phthalazinedione [
22]. The reaction of
31 with iodobenzene in the presence of triphenylarsine as a ligand and additionally sodium fluoride at room temperature led to 3% of the expected hydroarylation product
34, only, while 67% of
35 was formed as the product of a C-N cleavage reaction. Formally, the formation of
35 is the result of a 1,2-hydrazidoarylation on the primarily employed 1,3-cyclo-pentadiene (
Scheme 5).
Scheme 5.
Competition in stereoselectivity: hydroarylation vs. hydrazidoarylation.
Scheme 5.
Competition in stereoselectivity: hydroarylation vs. hydrazidoarylation.
The yield of the hydroarylation products under standard conditions is highly dependent on the rigid tricyclic system used: the PTAD cycloadduct of 1,3-cyclopentadiene
36 is best suited, and the yield drops by a factor of almost three in the case of the diazabicyclo[2.2.2]octene system
38, due to both the lower reactivity of the less strained double bond and the steric interactions with both CH-groups. When the C=C double bond is covered by a cyclopropane ring as in compound
40,
exo-attack is no longer feasible (
Scheme 6).
Scheme 6.
Hydroarylation of the cycloadducts of PTAD to cyclic dienes: dependence on the ring size.
Scheme 6.
Hydroarylation of the cycloadducts of PTAD to cyclic dienes: dependence on the ring size.
The key diazabicyclic systems become more flexible when a larger cycle is used, e.g. 1,3-cyclo-octadiene (1,3-COD). As an exception, in case of the PTAD cycloadduct
41 a twofold Heck reaction takes place in good yield to give
44 with an intermediate isomerization of the remaining
anti-Bredt double bond from
42 to
43. The structure of
44 was confirmed by an X-ray crystal structure analysis. Even under reductive conditions no hydroarylation product could be isolated. Therefore, in general the reductive elimination of hydridopalladium iodide to give
42 is apparently much faster than the reductive cleavage of the primarily formed carbopalladation product in the course of a hydroarylation reaction (
Scheme 7).
Scheme 7.
Mechanism of the hydroarylation of the PTAD cycloadduct of 1,3-cyclo-octadiene: ring flexibility leads to a twofold Heck reaction.
Scheme 7.
Mechanism of the hydroarylation of the PTAD cycloadduct of 1,3-cyclo-octadiene: ring flexibility leads to a twofold Heck reaction.
Inter- or intramolecular sequential insertion reactions of a carbopalladation product into a strained cyclopropane C-C σ-bond were unknown for a long time. The easily accessible
endo,
exo-bishomobarrelene
44 represents a
cis-allylcyclopropane model compound which can be attacked from one side only, due to the shielding by the second cyclopropane ring (
Scheme 8).
Scheme 8.
Mechanism of the palladium-catalyzed rearrangement of 45 under arylation.
Scheme 8.
Mechanism of the palladium-catalyzed rearrangement of 45 under arylation.
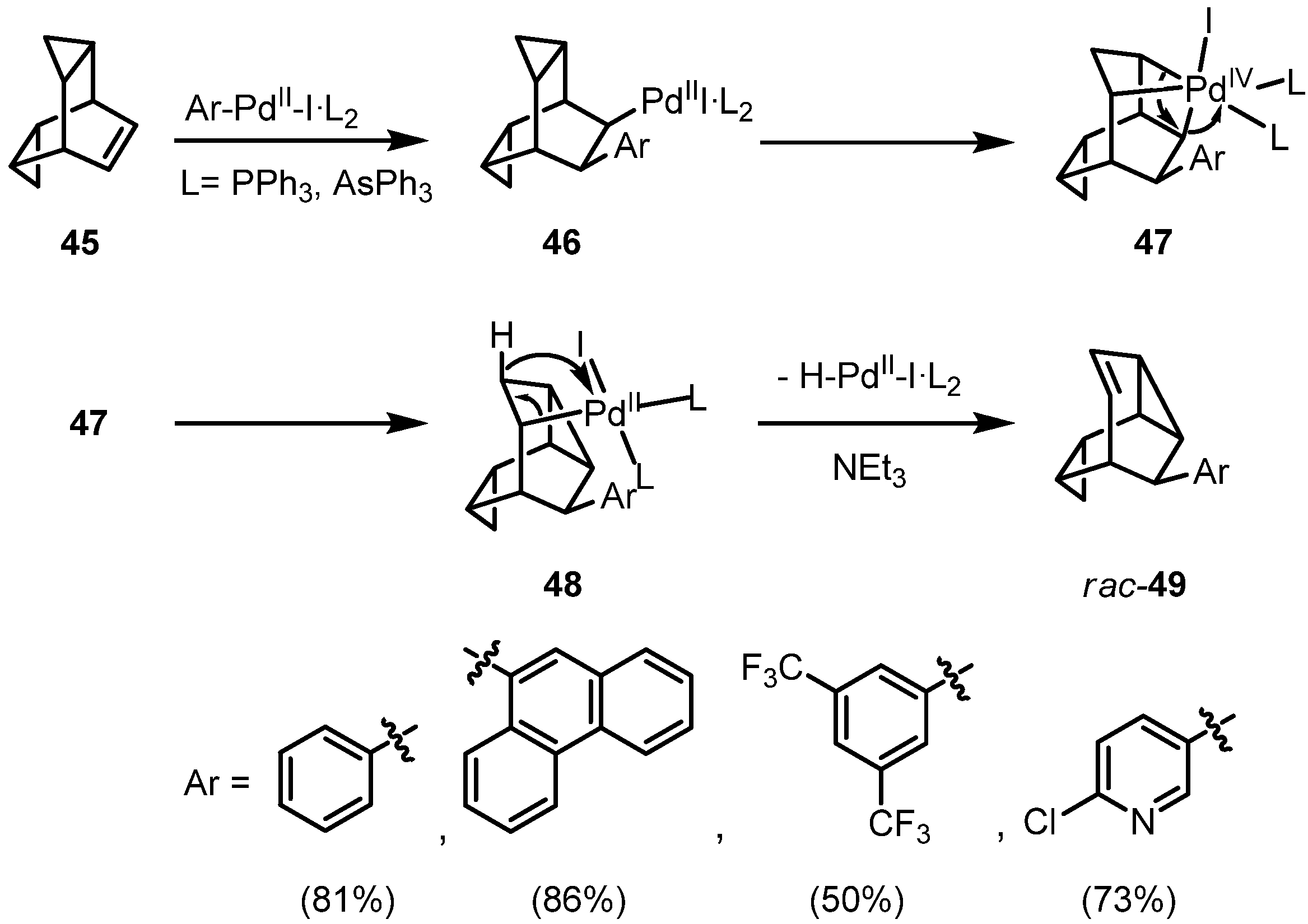
Treatment of
45 with iodobenzene under standard hydroarylation conditions surprisingly leads to the formation of 9-phenylmonohomosemibullvalene (
rac-49) in 81% yield; not even traces of a hydroarylation product was formed. In comparison, reaction of the less strained cyclobuta-annelated bicyclo[2.2.2]hexene again results in a reductive arylation. The palladium-catalyzed phenylation of an allylcyclopropane unit under sequential rearrangement is novel and may be classified as the first example of a π,σ domino-Heck reaction. We suggest the pathway outlined in
Scheme 8: after an initial
syn-addition of the Ph-Pd-I species to the C=C double bond, the palladium(II) substituent can interact with the Walsh-orbitals of the neighboring
endo-cyclopropane ring, leading by an oxidative addition from palladium(II) to palladium(IV). This highly strained octahedral complex can be stabilized by a reductive elimination to the σ-alkylpalladium(II) complex
48 under concomitant rearrangement. Compound
48 has the structural prerequisites for a final
syn-elimination of hydridopalladium iodide with formation of racemic
49. Addition of a chiral bisphosphine ligand such as (
R)-BINAP allows one to control seven chiral centers within a single operation [
23].





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






