Effects of P-Glycoprotein and Its Inhibitors on Apoptosis in K562 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Expression of P-gp in K562 Cells

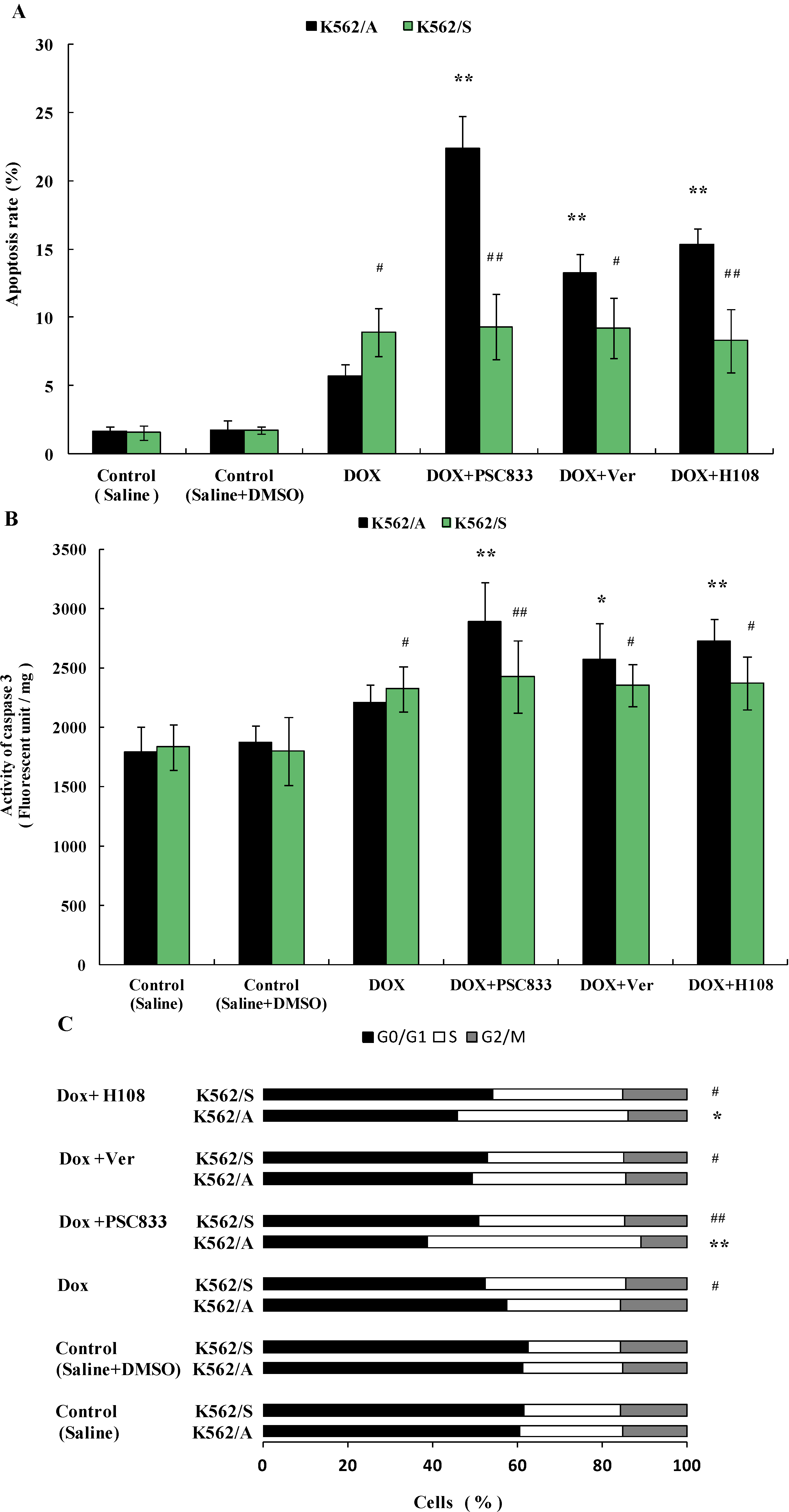
2.2. Apoptosis in K562 Cells Induced by Doxorubicin (Dox)

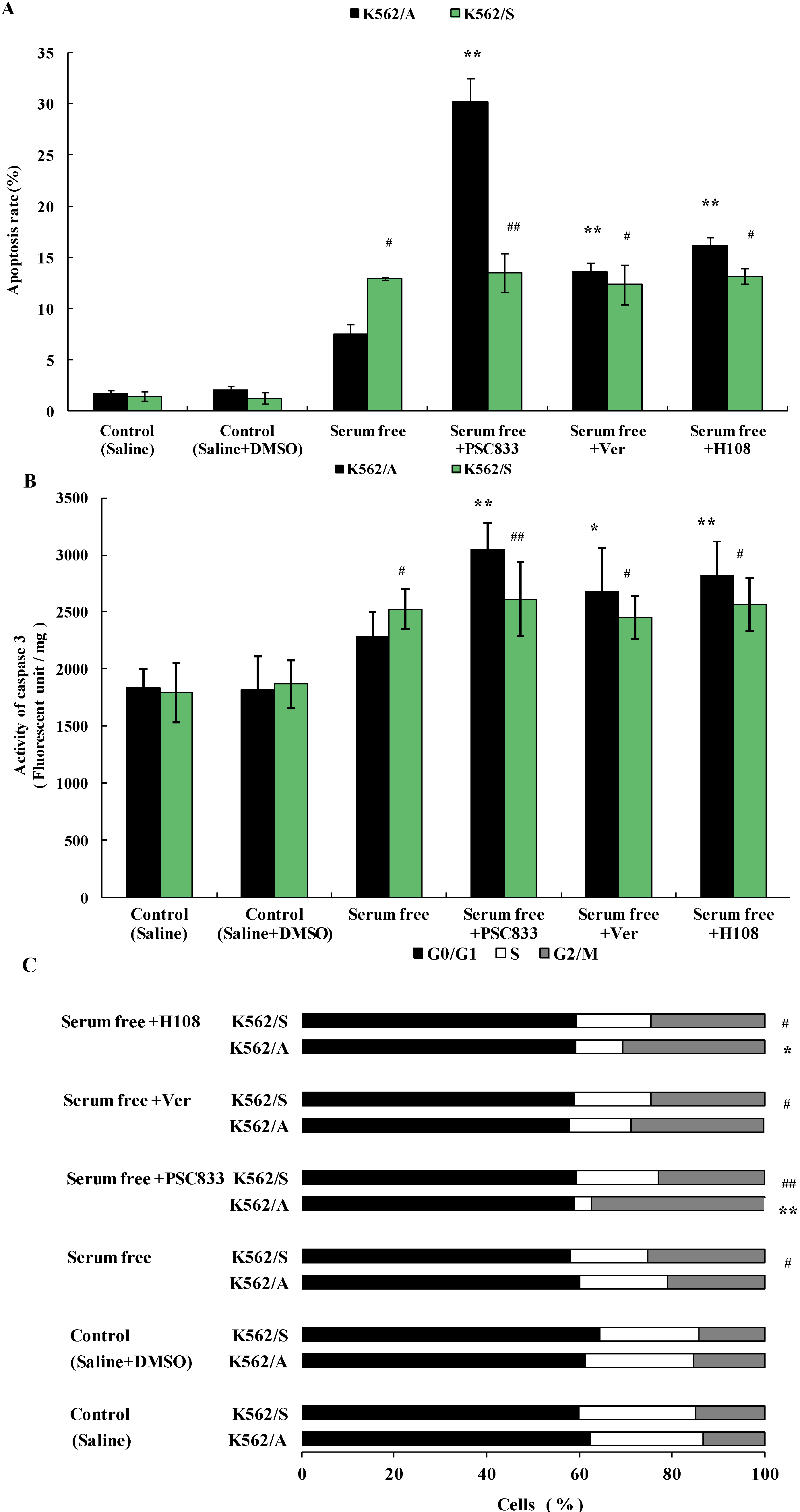
2.3. Apoptosis of K562 Cells Induced during Serum Deprivation

2.4. Effects of PSC833 in Viability of K562 Cells

2.5. PSC833-Induced Apoptosis of K562 Cells
2.6. Discussions
3. Experimental Section
3.1. Cell Culture
3.2. Western Blot Analysis of P-Glycoprotein Cells Culture
3.3. Apoptosis Induction
3.4. Apoptosis and Cell Cycle Analysed by Flow Cytometry
3.5. Caspase 3 Activity Determined by Immunoassay
3.6. MTT Assay of K562 Cells after PSC833 Treatment
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pérez-Tomás, R. Multidrug resistance: Retrospect and prospects in anti-cancer drug treatment. Curr. Med. Chem. 2006, 13, 1859–1876. [Google Scholar] [CrossRef]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef]
- Wang, L.; Meng, Q.; Wang, C.; Liu, Q.; Peng, J.; Huo, X.; Sun, H.; Ma, X.; Liu, K. Dioscin restores the activity of the anticancer agent adriamycin in multidrug-resistant human leukemia K562/adriamycin cells by down-regulating MDR1 via a mechanism involving NF-κB signaling inhibition. J. Nat. Prod. 2013, 76, 909–914. [Google Scholar] [CrossRef]
- Shen, F.; Chu, S.; Bence, AK.; Bailey, B.; Xue, X.; Erickson, PA.; Montrose, MH.; Beck, W.T.; Erickson, L.C. Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells. J. Pharmacol. Exp. Ther. 2008, 324, 95–102. [Google Scholar]
- Hannun, Y.A. Apoptosis and the dilemma of cancer chemotherapy. Blood 1997, 89, 1845–1853. [Google Scholar]
- Russo, A.; Terrasi, M.; Agnese, V.; Santini, D.; Bazan, V. Apoptosis a relevant tool for anticancer therapy. Ann. Oncol. 2006, 17, 115–123. [Google Scholar] [CrossRef]
- Ganguly, A.; Basu, S.; Banerjee, K.; Chakraborty, P.; Sarkar, A.; Chatterjee, M.; Chaudhuri, S.K. Redox active copper chelate overcomes multidrug resistance in T-lymphoblastic leukemia cell by triggering apoptosis. Mol. Biosyst. 2011, 7, 1701–1712. [Google Scholar] [CrossRef]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef]
- Gupta, S.; Gollapudi, S. P-glycoprotein (MDR 1 gene product) in cells of the immune system. Its possible physiologic role and alteration in aging and human immunodeficiency virus-1 (HIV-1) infection. J. Clin. Immunol. 1993, 13, 289–301. [Google Scholar] [CrossRef]
- Myllynen, P.; Kummu, M.; Sieppi, E. ABCB1 and ABCG2 expression in the placenta and fetus: An interspecies comparison. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1385–1398. [Google Scholar] [CrossRef]
- Mizutani, T.; Masuda, M.; Nakai, E.; Furumiya, K.; Togawa, H.; Nakamura, Y.; Kawai, Y.; Nakahira, K.; Shinkai, S.; Takahashi, K. Genuine functions of P-glycoprotein (ABCB1). Curr. Drug. Metab. 2008, 9, 167–174. [Google Scholar] [CrossRef]
- Sui, H.; Fan, Z.Z.; Li, Q. Signal transduction pathways and transcriptional mechanisms of ABCB1/Pgp-mediated multiple drug resistance in human cancer cells. J. Int. Med. Res. 2012, 40, 426–435. [Google Scholar] [CrossRef]
- Viale, M.; Cordazzo, C.; de Totero, D.; Budriesi, R.; Rosano, C.; Leoni, A.; Ioan, P.; Aiello, C.; Croce, M.; Andreani, A.; et al. Inhibition of MDR1 activity and induction of apoptosis by analogues of nifedipine and diltiazem: an in vitro analysis. Investig. New Drugs 2011, 29, 98–109. [Google Scholar] [CrossRef]
- Pallis, M.; Russell, N. P-glycoprotein plays a drug-efflux-independent role in augmenting cell survival in acute myeloblastic leukemia and is associated with modulation of a sphingomyelin-ceramide apoptotic pathway. Blood 2000, 95, 2897–2904. [Google Scholar]
- Turella, P.; Filomeni, G.; Dupuis, M.L.; Ciriolo, M.R.; Molinari, A.; de Maria, F.; Tombesi, M.; Cianfriglia, M.; Federici, G.; Ricci, G.; et al. A strong glutathione S-transferase inhibitor overcomes the P-glycoprotein-mediated resistance in tumor cells. 6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio) hexanol (NBDHEX) triggers a caspase-dependent apoptosis in MDR1-expressing leukemia cells. J. Biol. Chem. 2006, 281, 23725–23732. [Google Scholar] [CrossRef]
- Ruefli, A.A.; Smyth, M.J.; Johnstone, R.W. HMBA induces activation of a caspase-independent cell death pathwayto overcome P-glycoprotein-mediated multidrug resistance. Blood 2000, 95, 2378–2385. [Google Scholar]
- Wu, Q.L.; Wu, X.P.; Liang, Y.J.; Chen, L.M.; Ding, Y.; Fu, L.W. P-glycoprotein is not involved in pathway of anti-Fas/Fas-induced apoptosis in KBv200 cells. World J. Gastroenterol. 2005, 11, 3544–3548. [Google Scholar]
- Saraswathy, M.; Gong, S. Different strategies to overcome multidrug resistance in cancer. Biotechnol. Adv. 2013, 31, 1397–1407. [Google Scholar] [CrossRef]
- Darby, R.A.; Callaghan, R.; McMahon, R.M. P-glycoprotein inhibition the past, the present and the future. Curr. Drug Metab. 2011, 12, 722–731. [Google Scholar] [CrossRef]
- White, Y.; Hamada, T.; Yoshimitsu, M.; Nakashima, M.; Hachiman, M.; Kozako, T.; Matsushita, K.; Uozumi, K.; Suzuki, S.; Kofune, H.; et al. Novel cytotoxic isolated from Jamaican Hyptisverticillatajacq induces apoptosis and overcomes multidrug resistance. Anticancer Res. 2011, 31, 4251–4257. [Google Scholar]
- Robinson, L.J.; Roberts, W.K.; Ling, T.T.; Lamming, D.; Sternberg, S.S.; Roepe, P.D. Human MDR 1 protein overexpression delays the apoptotic cascade in Chinese hamster ovary fibroblasts. Biochemistry 1997, 36, 11169–11178. [Google Scholar] [CrossRef]
- Fantappiè, O.; Solazzo, M.; Lasagna, N.; Platini, F.; Tessitore, L.; Mazzanti, R. P-glycoprotein mediates celecoxib-induced apoptosis in multiple drug-resistant cell lines. Cancer Res. 2007, 67, 4915–4923. [Google Scholar] [CrossRef]
- Karwatsky, J.; Lincoln, M.C.; Georges, E. A mechanism for P-glycoprotein-mediated apoptosis as revealed by verapamil hypersensitivity. Biochemistry 2003, 42, 12163–12173. [Google Scholar] [CrossRef]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef]
- Hensley, P.; Mishra, M.; Kyprianou, N. Targeting caspases in cancer therapeutics. Biol. Chem. 2013, 394, 831–843. [Google Scholar]
- Mazumder, S.; Plesca, D.; Almasan, A. Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. Methods Mol. Biol. 2008, 414, 13–21. [Google Scholar]
- Kim, TH.; Shin, Y.J.; Won, A.J.; Lee, B.M.; Choi, W.S.; Jung, J.H.; Chung, H.Y.; Kim, H.S. Resveratrol enhances chemosensitivity of doxorubicin in multidrug-resistant human breast cancer cells via increased cellular influx of doxorubicin. Biochim. Biophys. Acta 2013, 1840, 615–625. [Google Scholar]
- O’Brien, M.M.; Lacayo, N.J.; Lum, B.L.; Kshirsagar, S.; Buck, S.; Ravindranath, Y.; Bernstein, M.; Weinstein, H.; Chang, M.N.; Arceci, R.J.; et al. Phase I study of valspodar (PSC-833) with mitoxantrone and etoposide in refractory and relapsed pediatric acute leukemia. A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2010, 54, 694–702. [Google Scholar] [CrossRef]
- Yusa, K.; Tsuruo, T. Reversal mechanism of multidrug resistance by verapamil. Direct binding of verapamil to P-glycoprotein on specific sites and transport of verapamil outward across the plasma membrane of K562/ADM cells. Cancer Res. 1989, 49, 5002–5006. [Google Scholar]
- Yang, Z.Y.; Liu, G.Q.; Huang, W.L. Effects of novel tetrahydroisoquinoline derivative H108 on activity of P-glycoprotein vitro and injury of PC12 cells. Chin. J. Clin. Pharmacol. Ther. 2004, 9, 275–280. [Google Scholar]
- Zhang, M.H.; Hu, Y.D.; Xu, Y.; Xiao, Y.; Luo, Y.; Song, Z.C.; Zhou, J. Human mesenchymal stem cells enhance autophagy of lung carcinoma cells against apoptosis during serum deprivation. Int. J. Oncol. 2013, 42, 1390–1398. [Google Scholar]
- Smyth, M.J.; Krasovskis, E.; Sutton, V.R.; Johnstone, R.W. The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 7024–7029. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Cretney, E.; Smyth, M.J. P-glycoprotein protects leukemia cells against caspase-dependent, but not caspase-independent, cell death. Blood 1999, 93, 1075–1085. [Google Scholar]
- Gibalová, L.; Sereš, M.; Rusnák, A.; Ditte, P.; Labudová, M.; Uhrík, B.; Pastorek, J.; Sedlák, J.; Breier, A.; Sulová, Z. P-glycoprotein depresses cisplatin sensitivity in L1210 cells by inhibiting cisplatin-induced caspase-3 activation. Toxicol. In Vitro 2012, 26, 435–444. [Google Scholar] [CrossRef]
- Rumjanek, V.M.; Trindade, G.S.; Wagner-Souza, K.; de-Oliveira, M.C.; Marques-Santos, L.F.; Maia, R.C.; Capella, M.A. Multidrug resistance in tumour cells. Characterization of the multidrug resistant cell line K562-Lucena 1. An. Acad. Bras. Ciênc. 2001, 73, 57–69. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zu, Y.; Yang, Z.; Tang, S.; Han, Y.; Ma, J. Effects of P-Glycoprotein and Its Inhibitors on Apoptosis in K562 Cells. Molecules 2014, 19, 13061-13075. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules190913061
Zu Y, Yang Z, Tang S, Han Y, Ma J. Effects of P-Glycoprotein and Its Inhibitors on Apoptosis in K562 Cells. Molecules. 2014; 19(9):13061-13075. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules190913061
Chicago/Turabian StyleZu, Yaqiong, Zhiyong Yang, Songshan Tang, Ying Han, and Jun Ma. 2014. "Effects of P-Glycoprotein and Its Inhibitors on Apoptosis in K562 Cells" Molecules 19, no. 9: 13061-13075. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules190913061