
Imaging the Ultrafast Photoelectron Transfer Process in Alizarin-TiO2

Abstract
:1. Introduction
2. Theoretical Framework
2.1. Static Calculations

2.2. Dynamical Calculations
2.2.1. A Single Active Electron Model
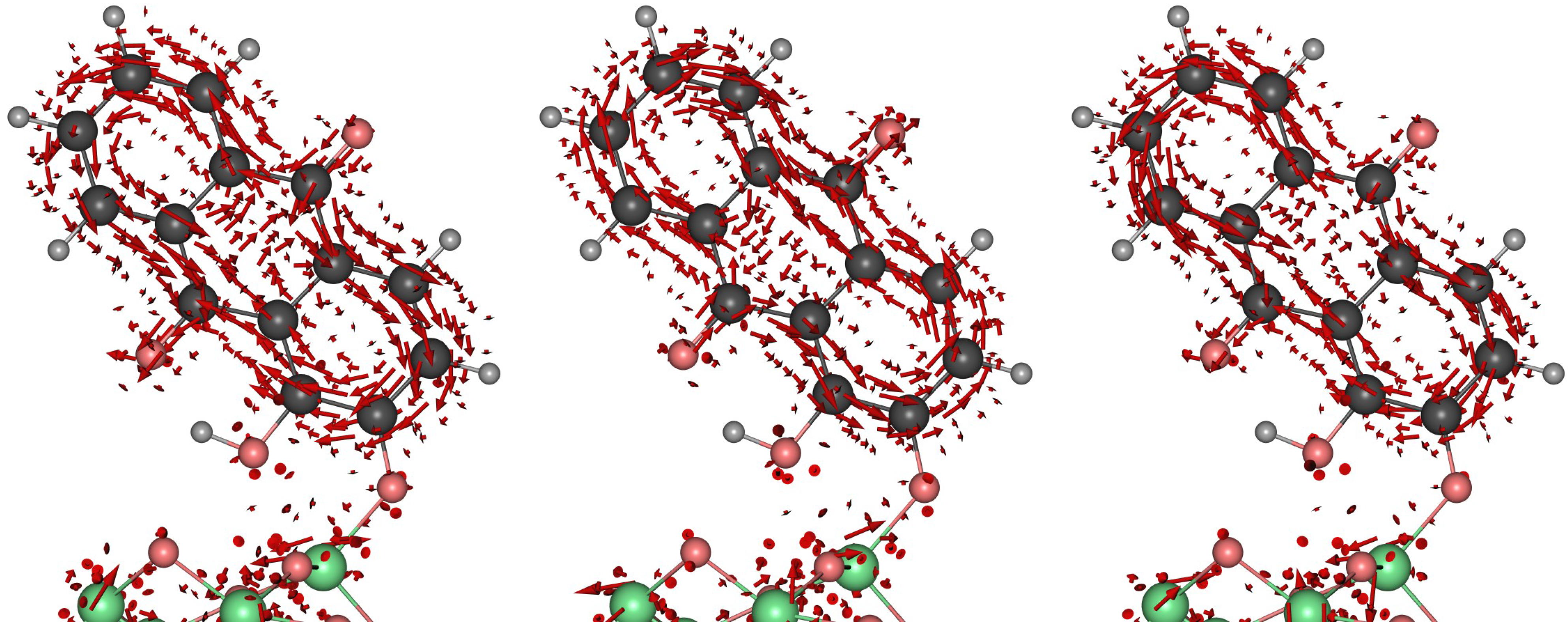
2.2.2. An Electronic Flux Analysis Toolset
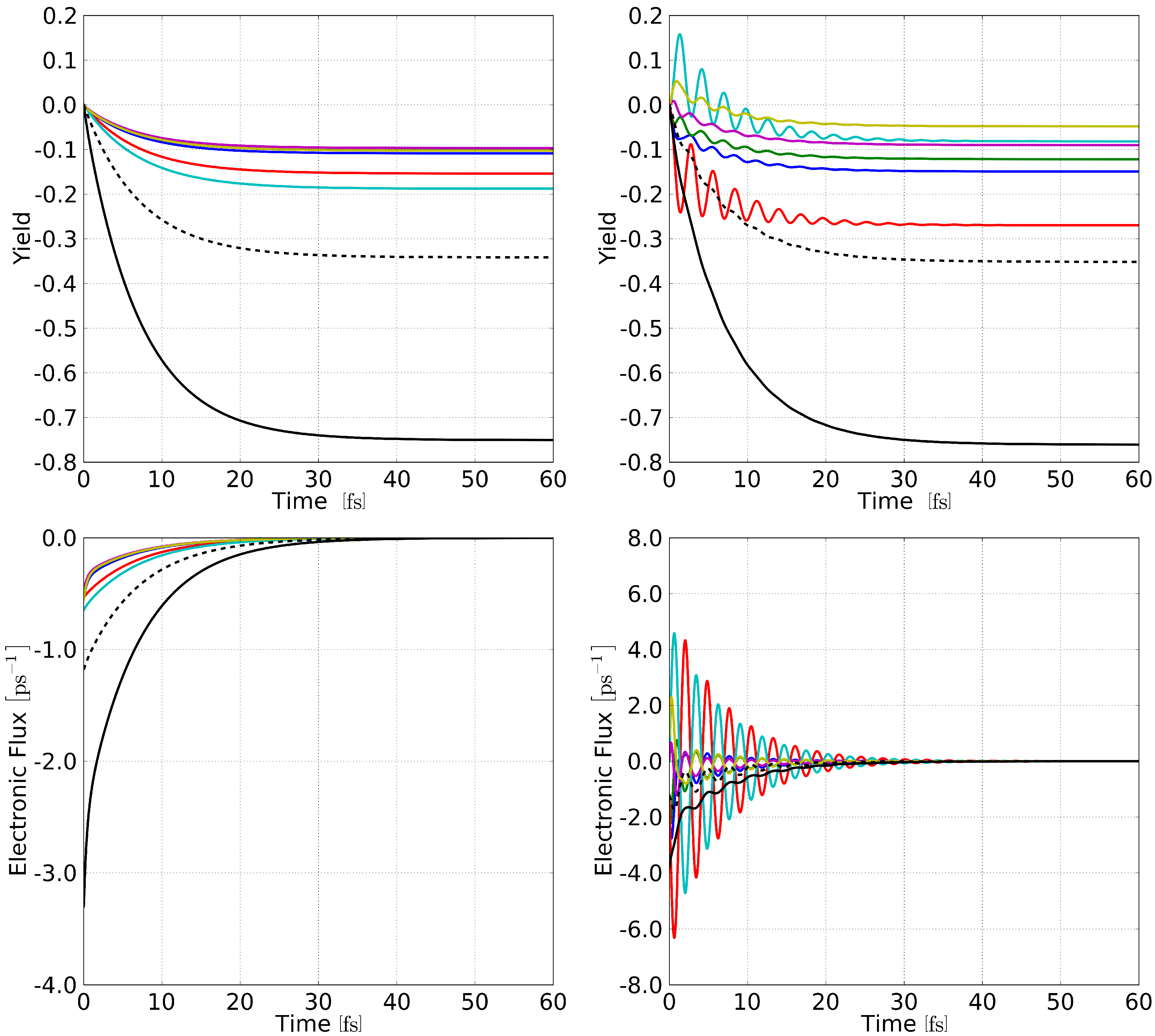
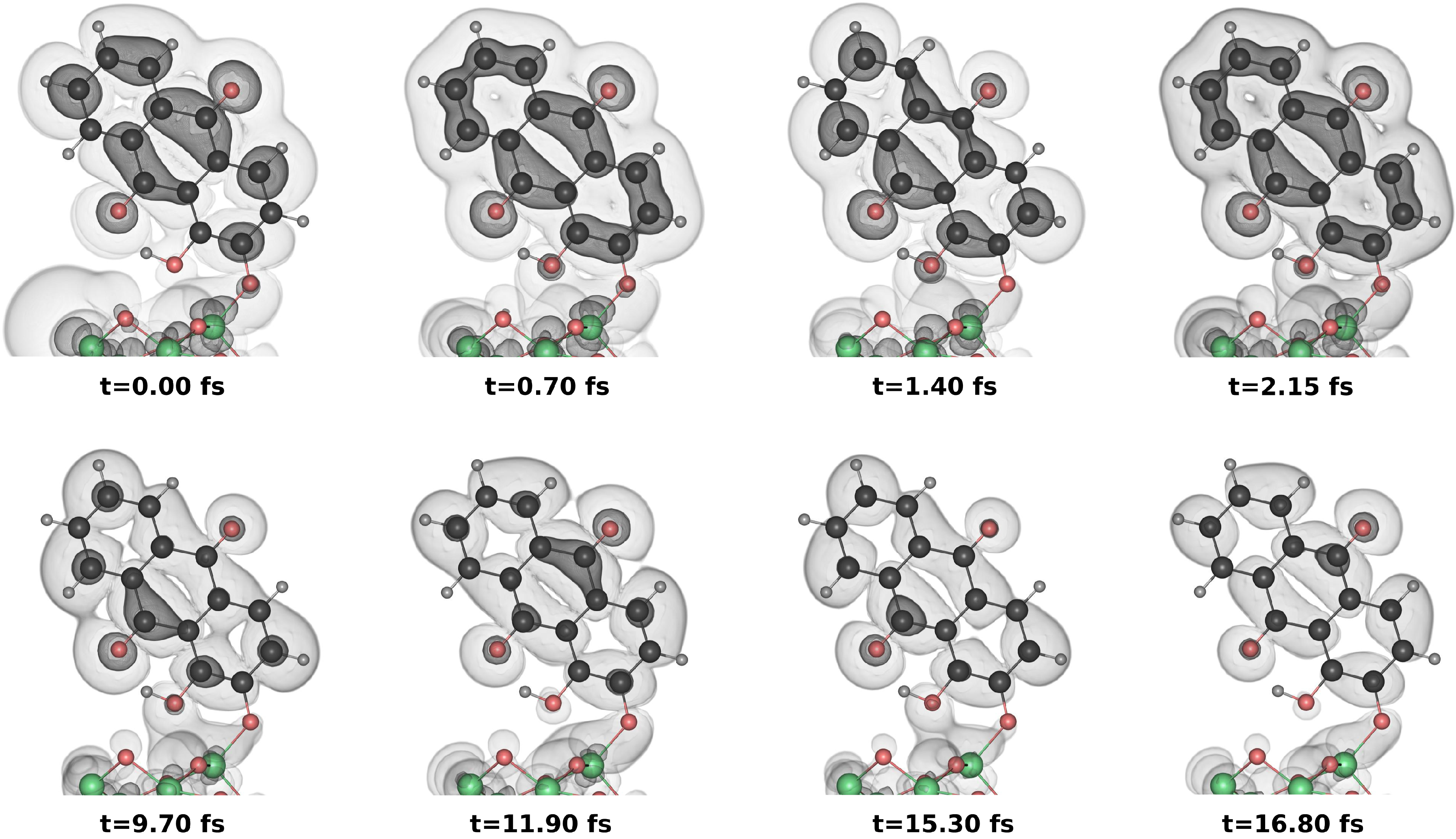
3. Results and Discussion
3.1. Static Considerations

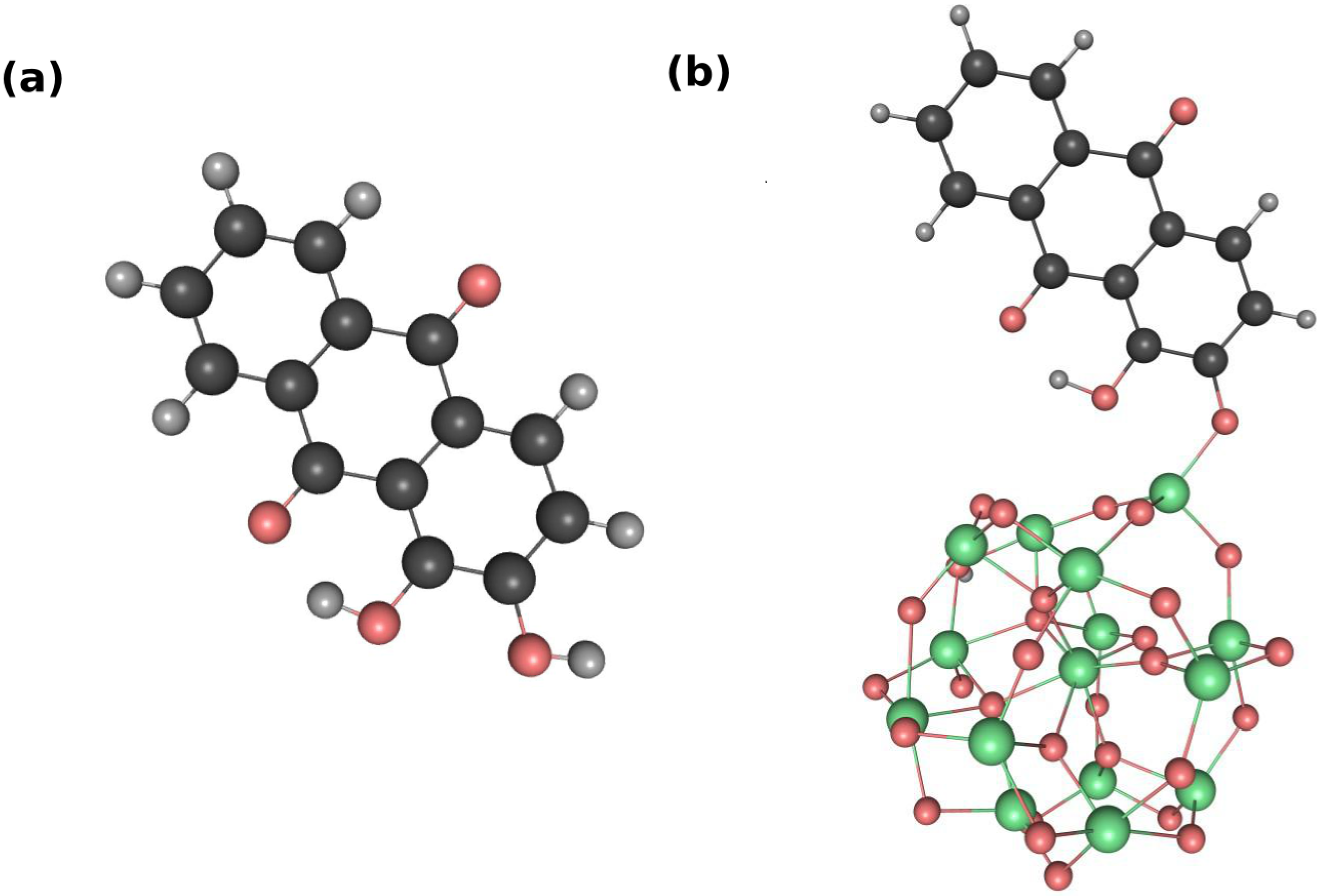{kind=link}
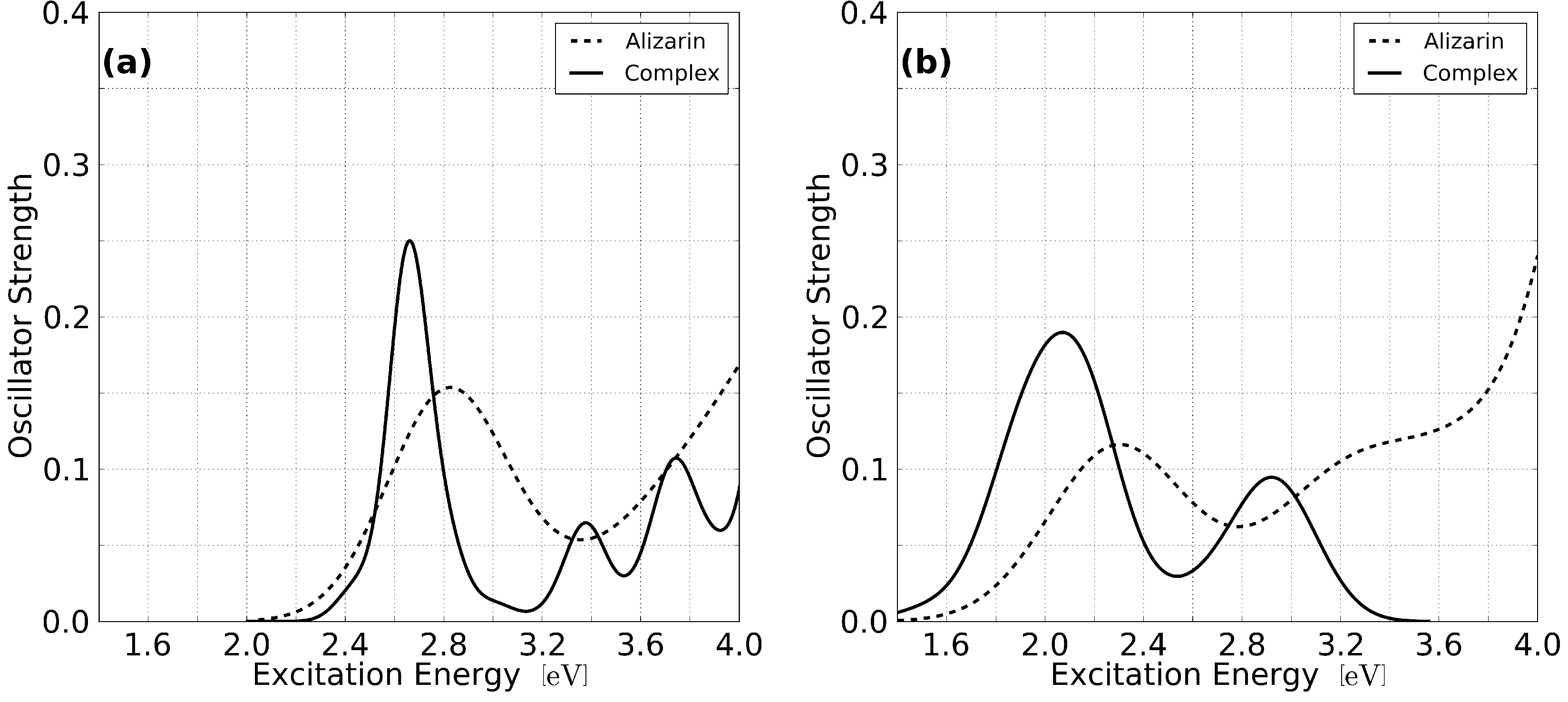{kind=link}
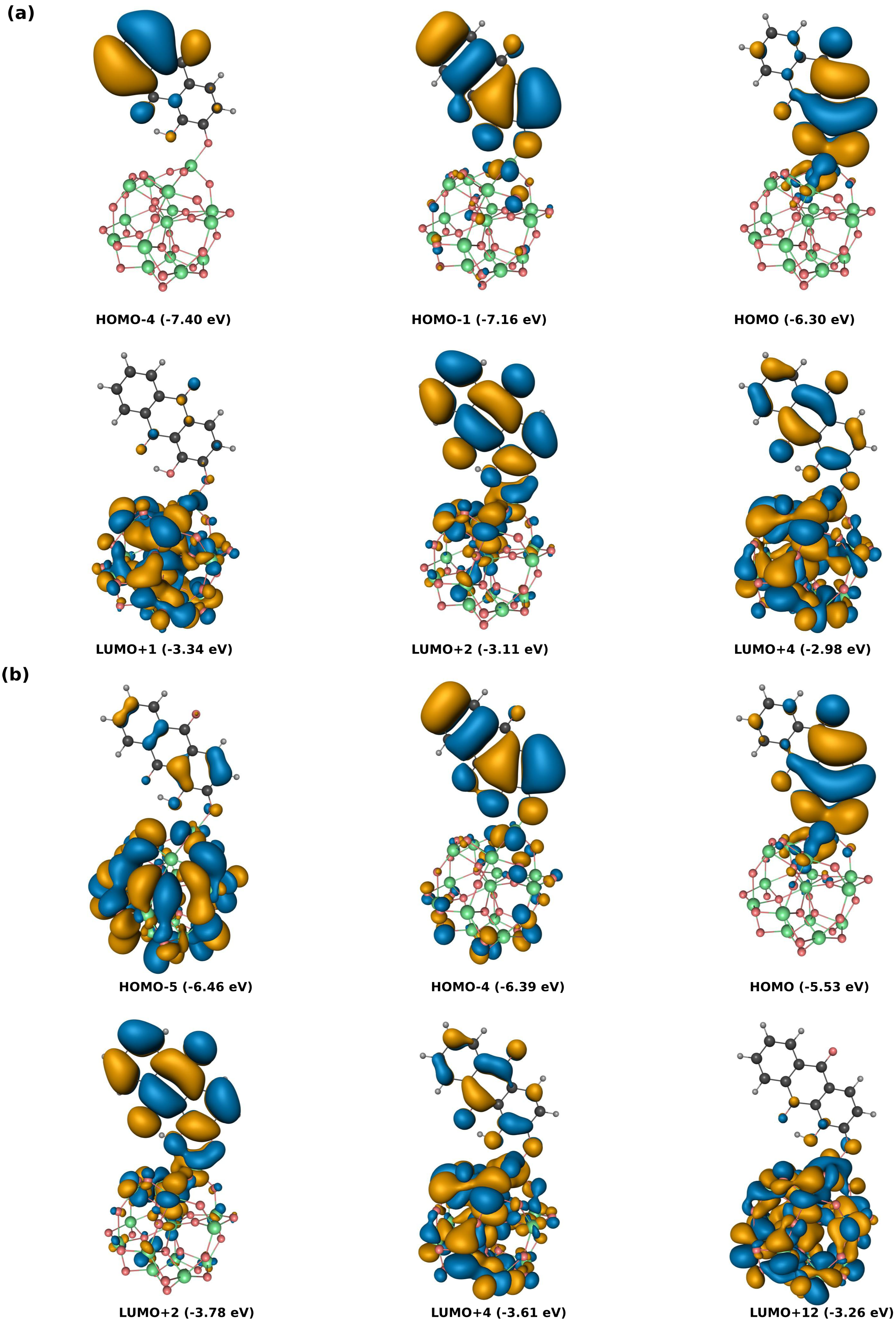{kind=link}
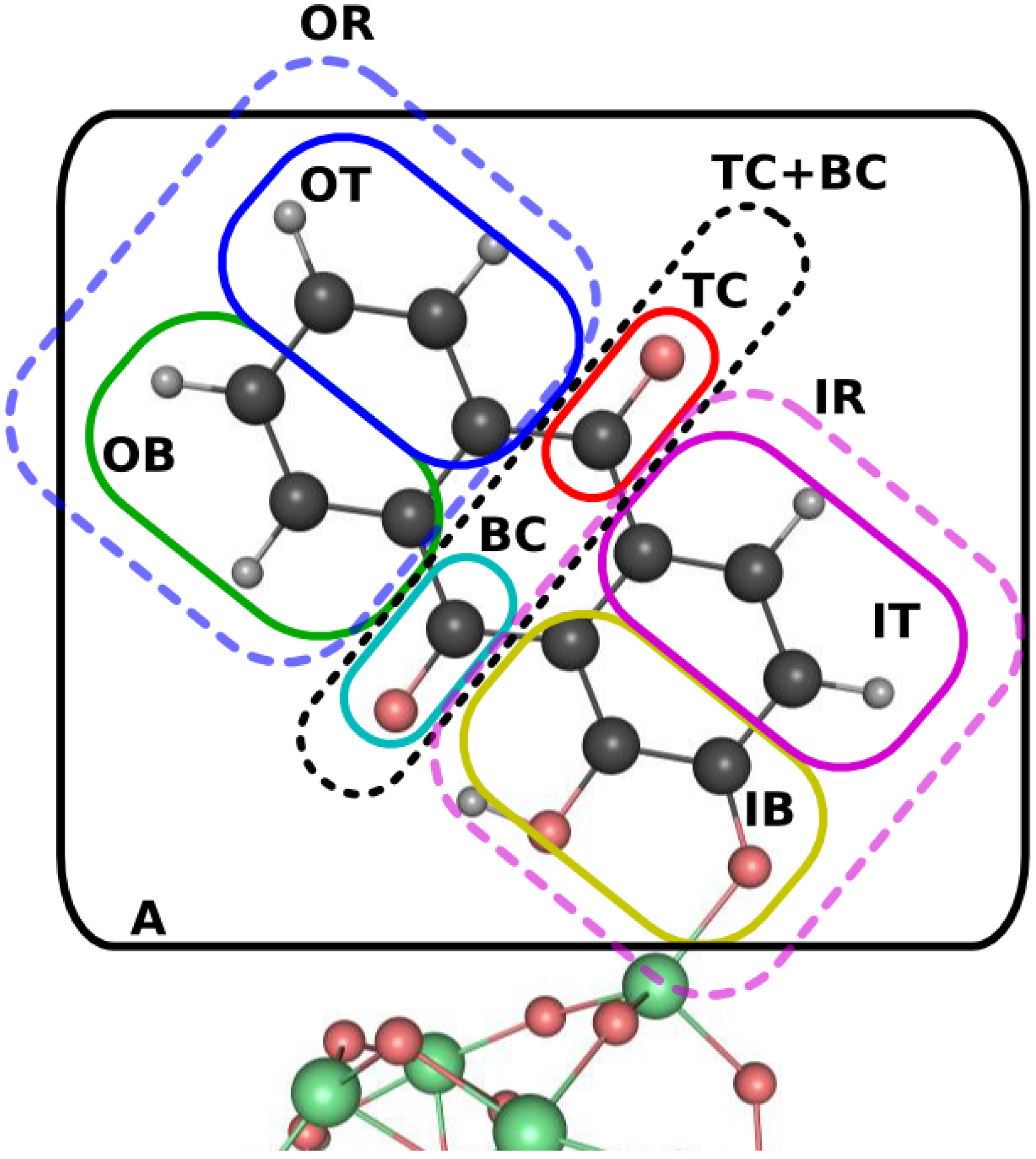{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alizarin | Alizarin-(TiO) | ||||
|---|---|---|---|---|---|
| Calc. | Exp. [14] | MO (%) | Calc. | Exp. | MO (%) |
| 2.82 | 2.88 | HOMO→LUMO (97.7) | 2.66 3.35 | 2.47 3.54 | HOMO→LUMO+2 (95.5) |
| HOMO-1→LUMO+1 (35.3) | |||||
| HOMO-1→LUMO+2 (25.8) | |||||
| HOMO-1→LUMO+5 (6.9) | |||||
| HOMO→LUMO+4 (11.7) | |||||
| 3.74 | 3.54 | HOMO-4→LUMO+2 (43.9) | |||
| HOMO-4→LUMO+1 (9.4) | |||||
| eHOMO-4→LUMO+5 (9.5) | |||||
| HOMO→LUMO+20 (7.8) | |||||
| HOMO→LUMO+22 (9.7) | |||||
| 2.29 3.28 | 2.88 3.82 | HOMO→LUMO (95.1) HOMO-4→LUMO (58.3) HOMO→LUMO+1 (35.0) | 2.16 | 2.47 | HOMO→LUMO+2 (25.6) |
| HOMO→LUMO+4 (11.3) | |||||
| HOMO→LUMO+11 (17.6) | |||||
| HOMO→LUMO+12 (24.0) | |||||
| 2.91 | 3.54 | HOMO-5→LUMO+3 (18.9) | |||
| HOMO-6→LUMO+2 (7.2) | |||||
| HOMO-4→LUMO+2 (10.7) | |||||
| HOMO-6→LUMO+4 (6.5) | |||||
| HOMO-4→LUMO+7 (11.7) | |||||
| HOMO→LUMO+26 (17.7) | |||||


3.2. Dynamical Considerations




4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Grätzel, M. Photoelectrochemical Cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Duncan, W.; Prezhdo, O. Theoretical studies of photoinduced electron transfer in dye-sensitized TiO2. Annu. Rev. Phys. Chem. 2007, 58, 143–184. [Google Scholar] [CrossRef] [PubMed]
- Yella, A.; Mai, C.L.; Zakeeruddin, S.M.; Chang, S.N.; Hsieh, C.H.; Yeh, C.Y.; Grätzel, M. Molecular Engineering of Push-Pull Porphyrin Dyes for Highly Efficient Dye-Sensitized Solar Cells: The Role of Benzene Spacers. Angew. Chem. Int. Ed. 2014, 53, 2973–2977. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Lenzmann, F.O.; Kroon, J.M.R. Recent Advances in Dye-Sensitized Solar Cells. Adv. OptoElectron. 2007, 2007. [Google Scholar] [CrossRef]
- Sánchez-de-Armas, R.; Oviedo, J.; San Miguel, M.A.; Fdez. Sanz, J. Direct vs. Indirect Mechanisms for Electron Injection in Dye-Sensitized Solar Cells. J. Phys. Chem. C 2011, 115, 11293–11301. [Google Scholar]
- Duncan, W.R.; Stier, W.M.; Prezhdo, O.V. Ab initio nonadiabatic molecular dynamics of the ultrafast electron injection across the alizarin-TiO2 interface. J. Am. Chem. Soc. 2005, 127, 7941–7951. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, M.; Li, J.; Pootrakulchote, N.; Alibabaei, L.; Ngoc-le, C.; Decoppet, J.D.; Tsai, J.; Grätzel, C.; Wu, C.; et al. Highly Efficient Light-Harvesting Ruthenium Sensitizer for Thin-Film Dye-Sensitized Solar Cells. ACS Nano 2009, 3, 3103–3109. [Google Scholar] [CrossRef] [PubMed]
- Hardin, B.E.; Snaith, H.J.; McGehee, M.D. The renaissance of dye-sensitized solar cells. Nat. Photonics 2012, 6, 162–169. [Google Scholar] [CrossRef]
- Nawrocka, A.; Krawczyk, S. Electronic Excited State of Alizarin Dye Adsorbed on TiO2 Nanoparticles: A Study by Electroabsorption (Stark Effect) Spectroscopy. J. Phys. Chem. C 2008, 112, 10233–10241. [Google Scholar] [CrossRef]
- Sánchez-de-Armas, R.; San-Miguel, M.A.; Oviedo, J.; Fdez. Sanz, J. Direct vs. indirect mechanisms for electron injection in DSSC: Catechol and alizarin. Comput. Theor. Chem. 2011, 975, 99–105. [Google Scholar]
- Pastore, M.; Mosconi, E.; de Angelis, F.; Grätzel, M.A. Computational Investigation of Organic Dyes for Dye-Sensitized Solar Cells: Benchmark, Strategies, and Open Issues. J. Phys. Chem. C 2010, 114, 7205–7212. [Google Scholar] [CrossRef]
- Huber, R.; Spörlein, S.; Moser, J.E.; Grätzel, M.; Wachtveitl, J. The role of Surface States in the Ultrafast Photoinduced Electron Transfer from Sensitizing Dye Molecules to Semiconductor Colloids. J. Phys. Chem. B 2000, 104, 8995–9003. [Google Scholar] [CrossRef]
- Huber, R.; Moser, J.E.; Grätzel, M.; Wachtveitl, J. Real-Time Observation of Photoinduced Adiabatic Electron Transfer in Strongly Coupled Dye/Semiconductor Colloidal Systems with a 6 fs Time Constant. J. Phys. Chem. B 2002, 106, 6494–6499. [Google Scholar] [CrossRef]
- Carta, L.; Biczysko, M.; Bloino, J.; Licari, D.; Barone, V. Enviromental and complexation effects on the structures and spectroscopic signatures of organic pigments relevant to cultural heritage: The case of alizarin and alizarin–Mg(II)/Al(III) complexes. Phys. Chem. Chem. Phys. 2014, 16, 2897–2911. [Google Scholar] [CrossRef] [PubMed]
- Armat, A.; Miliani, C.; Romani, A.; Fantacci, S. DFT/DFT investigation on the UV-vis absorption and fluorescence properties of alizarin dye. Phys. Chem. Chem. Phys. 2015, 17, 6374–6382. [Google Scholar] [CrossRef] [PubMed]
- Manmeeta; Dhiraj, S.; Sharma, G.D.; Roy, M.S. Improved performance of oxidized Alizarin based Quasi solid state Dye. Res. J. Chem. Sci. 2012, 2, 61–71. [Google Scholar]
- Oviedo, M.B.; Zarate, X.; Negre, F.A.; Schott, E.; Arratia-Pérez, R.; Sánchez, C.G. Quantum Dynamical Simulations as a Tool for Predicting Photoinjection Mechanisms in Dye-Sensitized TiO2 Solar Cells. J. Phys. Chem. Lett. 2012, 3, 2548–2555. [Google Scholar] [CrossRef]
- Duncan, W.R.; Prezhdo, O.V. Electronic Structure and Spectra of Catechol and Alizarin in the Gas Phase and Attached to Titanium. J. Phys. Chem. B 2005, 109, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Duncan, W.R.; Craig, C.F.; Prezhdo, O.V. Time-Domain ab Initio Study of Charge Relaxation and Recombination in Dye-Sensitized TiO2. J. Am. Chem. Soc. 2007, 129, 8528–8543. [Google Scholar] [CrossRef] [PubMed]
- Duncan, W.R.; Prezhdo, O.V. Temperature Independence of the Photoinduced Electron Injection in Dye-Sensitized TiO2 Rationalized by Ab Initio Time-Domain Density Functional Theory. J. Am. Chem. Soc. 2008, 130, 9756–9762. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-de Armas, R.; Oviedo, J.; San-Miguel, M.A.; Fdez. Sanz, J. Real-Time TD-DFT Simulations in Dye Sensitized Solar Cells: The Electronic Absorption Spectrum of Alizarin Supported on TiO2 Nanoclusters. Chem. Theory Comput. 2010, 6, 2856–2865. [Google Scholar] [CrossRef]
- Rego, L.G.C.; Batista, V.S. Quantum Dynamics Simulations of Interfacial Electron Transfer in Sensitized TiO2 Semiconductors. J. Am. Chem. Soc. 2003, 125, 7989–7997. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kondov, I.; Wang, H.; Thoss, M. Theoretical Study of Photoinduced Electron-Transfer Processes in the Dye-Semiconductor System Alizarin-TiO2. J. Phys. Chem. C 2010, 114, 18481–18493. [Google Scholar] [CrossRef]
- Li, J.; Wang, H.; Persson, P.; Thoss, M. Photoinduced electron transfer processes in dye-semiconductor systems with different spacer groups. J. Chem. Phys. 2012, 137, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Hamad, S.; Catlow, C.R.A.; Woodley, S.M.; Lago, S.; Mejías, J.A. Structure and Stability of Small TiO2 Nanoparticles. J. Phys. Chem. B 2005, 109, 15741–15748. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.V.; Tvrdy, K.; Baker, D.R.; Radich, J.G. Beyond Photovoltaics: Semiconductor Nanoarchitectures for Liquid-Junction Solar Cells. Chem. Rev. 2010, 110, 6664–6688. [Google Scholar] [CrossRef] [PubMed]
- Rocca, D.; Gebauer, R.; de Angelis, F.; Nazeeruddin, M.; Baroni, S. Time-dependent density functional theory study of squaraine dye-sensitized solar cells. Chem. Phys. Lett. 2009, 475, 49–53. [Google Scholar] [CrossRef]
- Mosconi, E.; Jun-Ho, Y.; Kessler, F.; Gómez, C.J.; Zuccaccia, C.; Cinti, A.; Nazeeruddin, M.K.; Grätzel, M.; de Angelis, F. Cobalt Electrolyte/Dye Interactions in Dye-Sensitized Solar Cells: A Combined Computational and Experimental Study. J. Am. Chem. Soc. 2012, 134, 19438–19453. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, C.; Mosconi, E.; Pastore, M.; Ronca, E.; de Angelis, F. Adsorption of organic dyes on TiO2 surfaces in dye-sensitized solar cells: Interplay of theory and experiment. Phys. Chem. Chem. Phys. 2012, 14, 15963–15974. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1998, 38, 3098–3100. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations- potentials for the transition-metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Fu, H.; Cheng, Y.M.; Wu, K.L.; Ho, S.T.; Chi, Y.; Chou, P.T. Theoretical Study of N749 Dyes Anchoring on the (TiO2)28 Surface in DSSCs and Their Electronic Absorption Properties. J. Phys. Chem. C 2012, 116, 16338–16345. [Google Scholar] [CrossRef]
- Gross, E.K.U.; Kohn, W. Time-Dependent Density-Functional Theory. Adv. Quantum Chem. 1990, 21, 255–291. [Google Scholar]
- Meng, S.; Kaxiras, E. Real-time, local basis-set implementation of the time-dependent density functional for excited states dynamics simulations. J. Chem. Phys. 2008, 129, 054110. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.; Paulus, B.; Pérez-Torres, J.F.; Pohl, V. Electronic and nuclear flux densities in the H2 molecule. Phys. Rev. A 2014, 89, 052504. [Google Scholar]
- Krause, P.; Sonk, J.A.; Schlegel, H.B. Strong field ionization rates simulated with time-dependent configuration interaction and an absorbing potential. J. Chem. Phys. 2014, 140, 174113. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, E. Quantisierung als Eigenwertproblem (Vierte Mitteilung). Ann. Phys. (Leipzig) 1926, 81, 109–139. [Google Scholar] [CrossRef]
- Freedman, T.B.; Gao, X.; Shih, M.L.; Nafie, L.A. Electron Transition Current Density in Molecules. 2. Ab Initio Calculations for Electronic Transitions in Ethylene and Formaldehyde. J. Phys. Chem. A 1998, 102, 3352–3357. [Google Scholar]
- Okuyama, M.; Takatsuka, K. Electron flux in molecules induced by nuclear motion. Chem. Phys. Lett. 2009, 476, 109–115. [Google Scholar] [CrossRef]
- Diestler, D.J.; Kenfack, A.; Manz, J.; Paulus, B.; Pérez-Torres, J.F.; Pohl, V. Computation of the Electronic Flux Density in the Born-Oppenheimer Approximation. J. Phys. Chem. A 2013, 117, 8519–8527. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, J.F. Electronic flux densities in vibrating in terms of vibronic eigenstates. Phys. Rev. A 2013, 87, 062512. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Handgraaf, J.W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.; Pohl, V.; Schild, A. Orbkit: A Toolbox for Post-Processing Quantum Chemical Wavefunction Data. 2014. Available online: http://sourceforge.net/p/orbkit (accessed on 9 July 2015).
- Stalling, D.; Westerhoff, M.; Hege, H.C. Amira: A Highly Interactive System for Visual Data Analysis. In Visualization Handbook; Hansen, C.D., Johnson, C.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Chapter 8; pp. 749–767. [Google Scholar]
- Kožišek, J.; Breza, M.; Ulický, L. Aromatic Character of Anthraquinone Derivates. Chem. Pap. 1993, 47, 34–37. [Google Scholar]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez, T.; Hermann, G.; Zarate, X.; Pérez-Torres, J.F.; Tremblay, J.C. Imaging the Ultrafast Photoelectron Transfer Process in Alizarin-TiO2. Molecules 2015, 20, 13830-13853. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules200813830
Gomez T, Hermann G, Zarate X, Pérez-Torres JF, Tremblay JC. Imaging the Ultrafast Photoelectron Transfer Process in Alizarin-TiO2. Molecules. 2015; 20(8):13830-13853. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules200813830
Chicago/Turabian StyleGomez, Tatiana, Gunter Hermann, Ximena Zarate, Jhon Fredy Pérez-Torres, and Jean Christophe Tremblay. 2015. "Imaging the Ultrafast Photoelectron Transfer Process in Alizarin-TiO2" Molecules 20, no. 8: 13830-13853. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules200813830