
Novel Natural Product- and Privileged Scaffold-Based Tubulin Inhibitors Targeting the Colchicine Binding Site

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
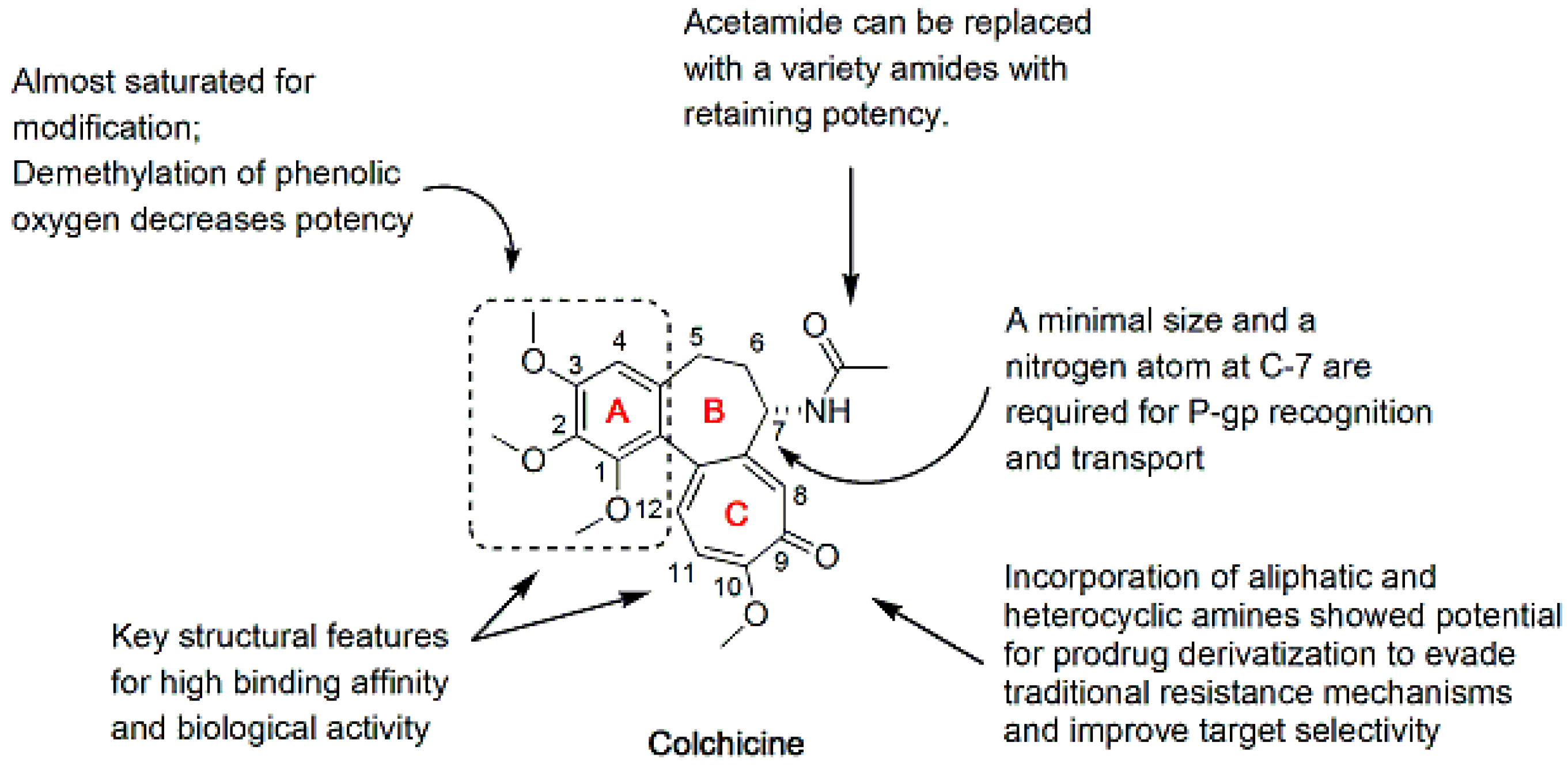
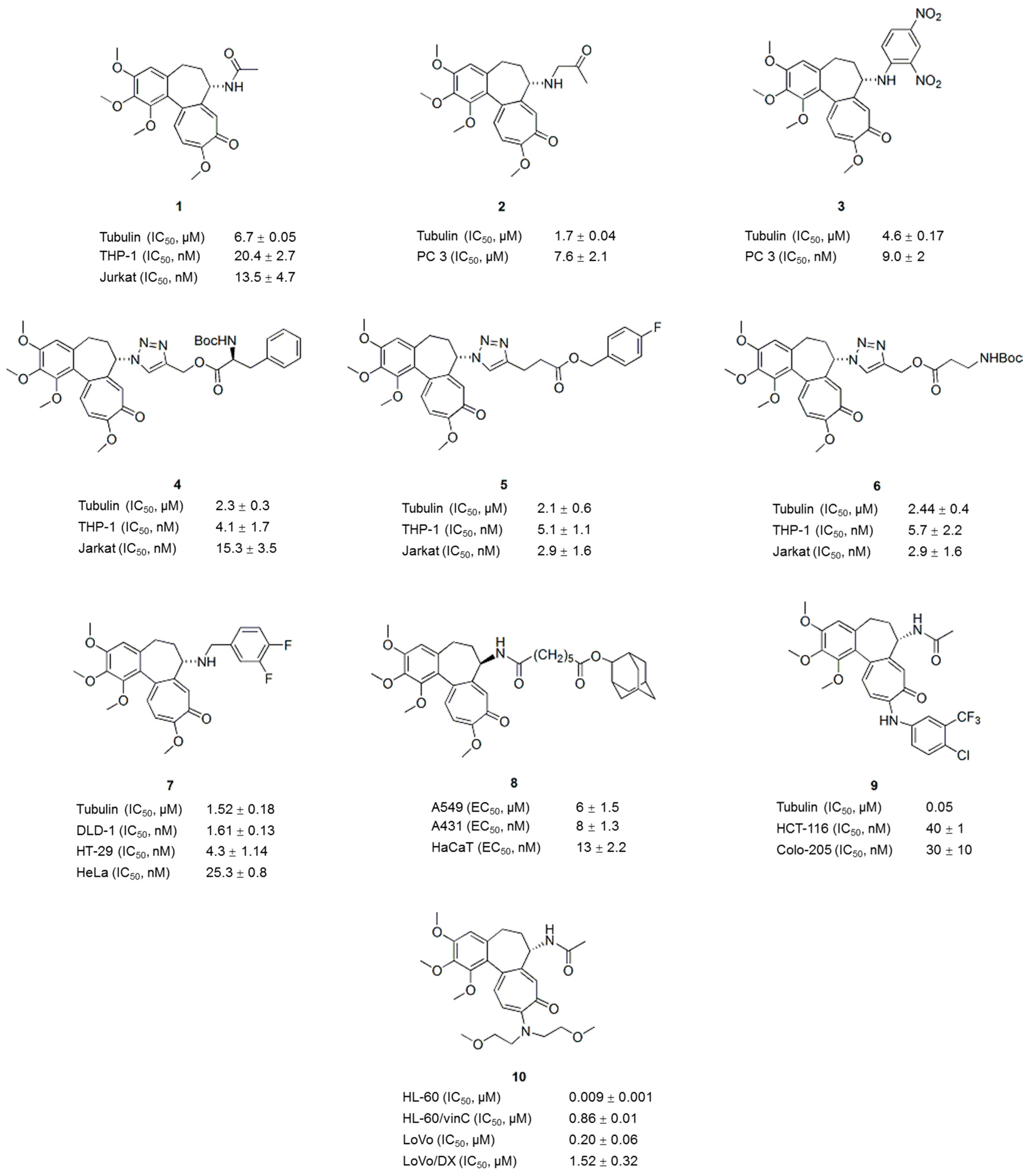
2. Colchicine Analogues
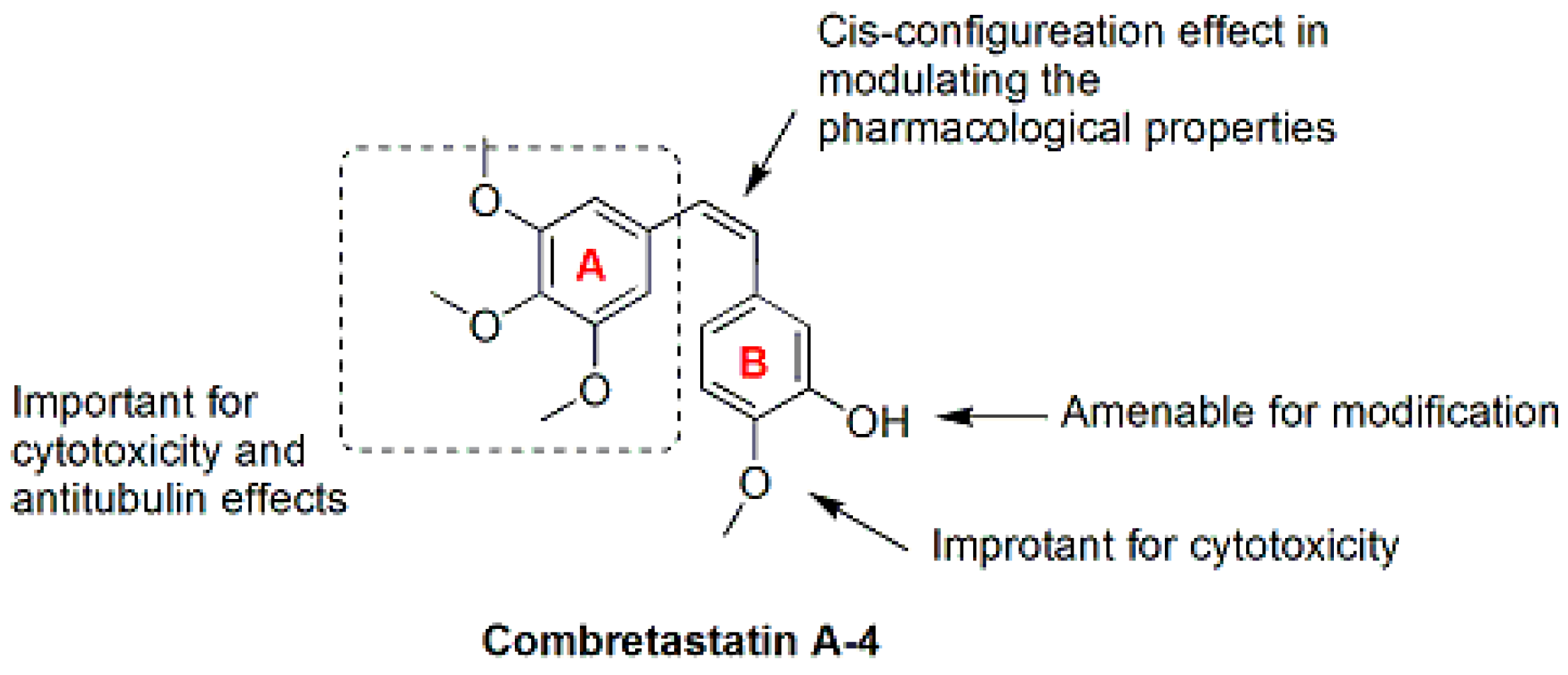
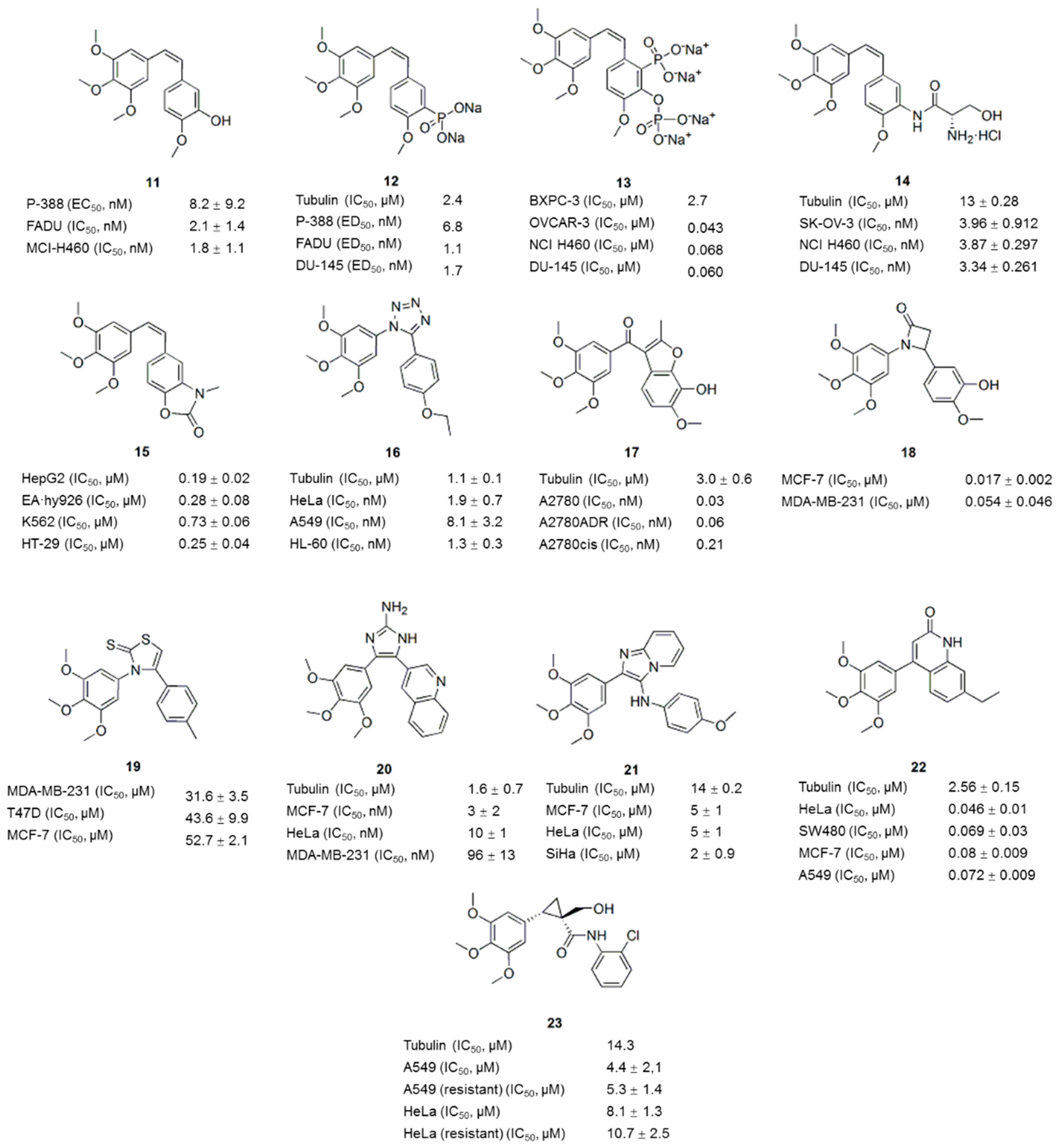
3. Combretastatin A-4 Analogues
3.1. B-Ring-Modified Analogues
3.2. Bridge-Modified Analogues
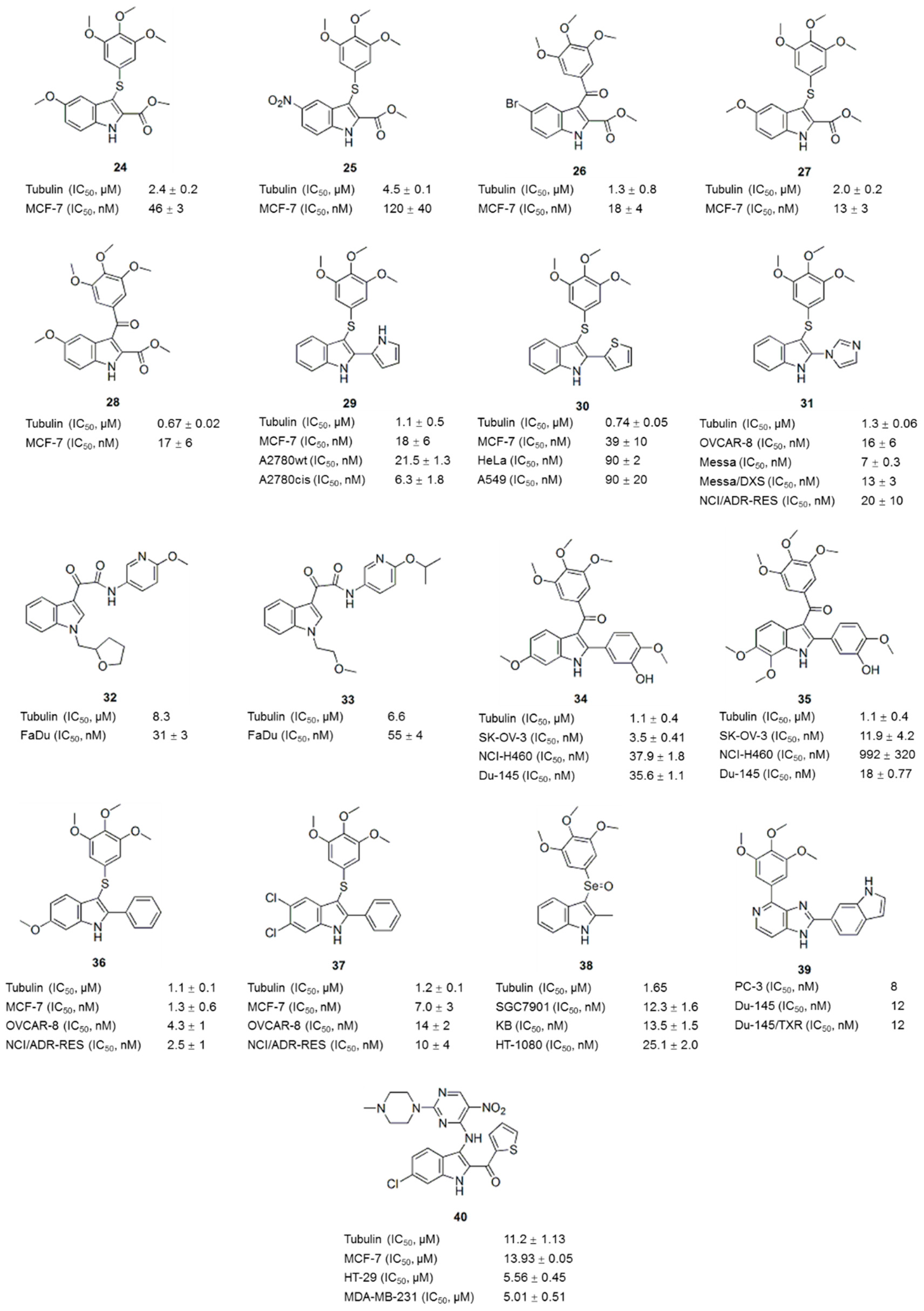
4. Compounds with an Indole Nucleus
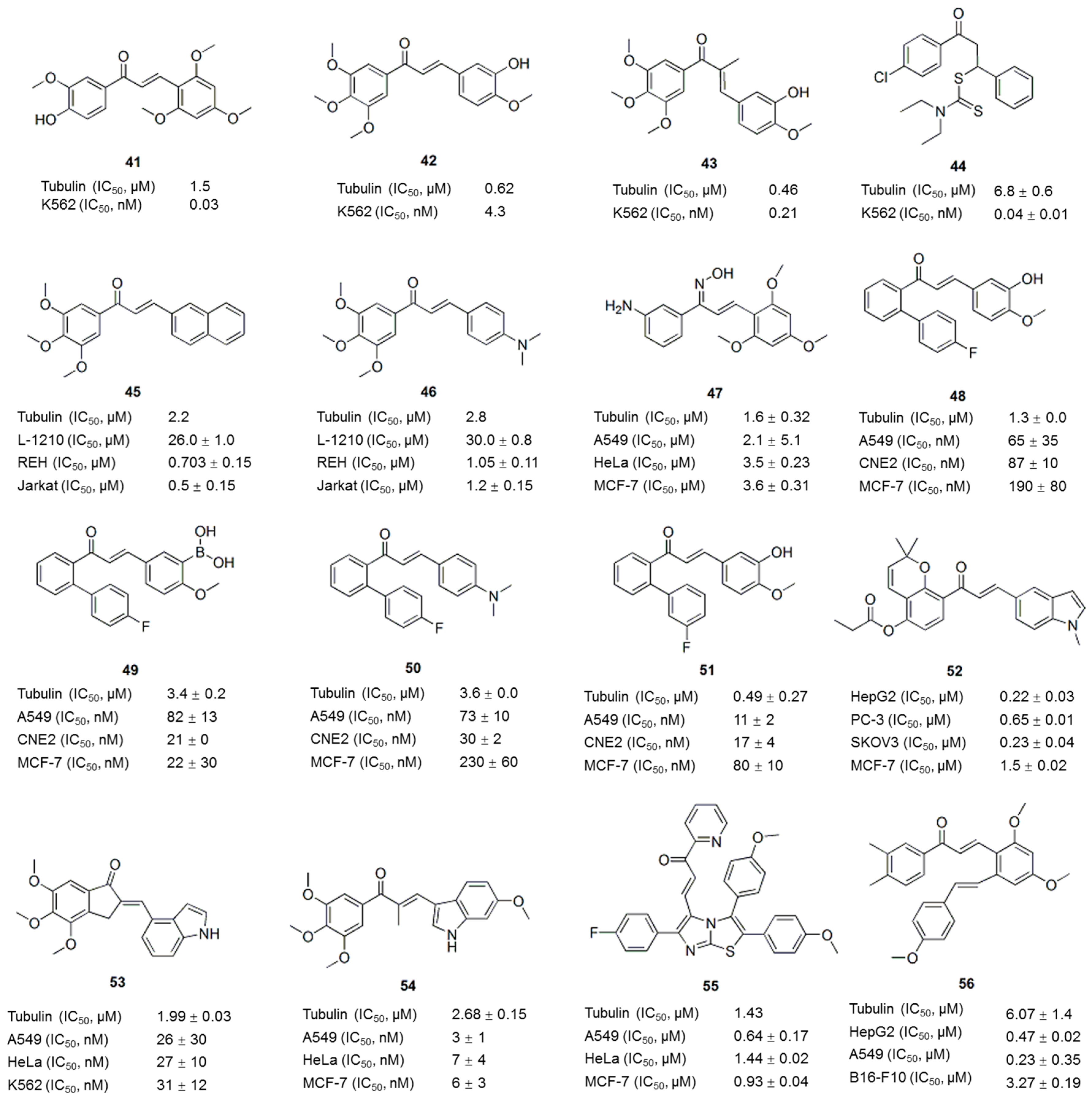
5. Chalcone Analogues
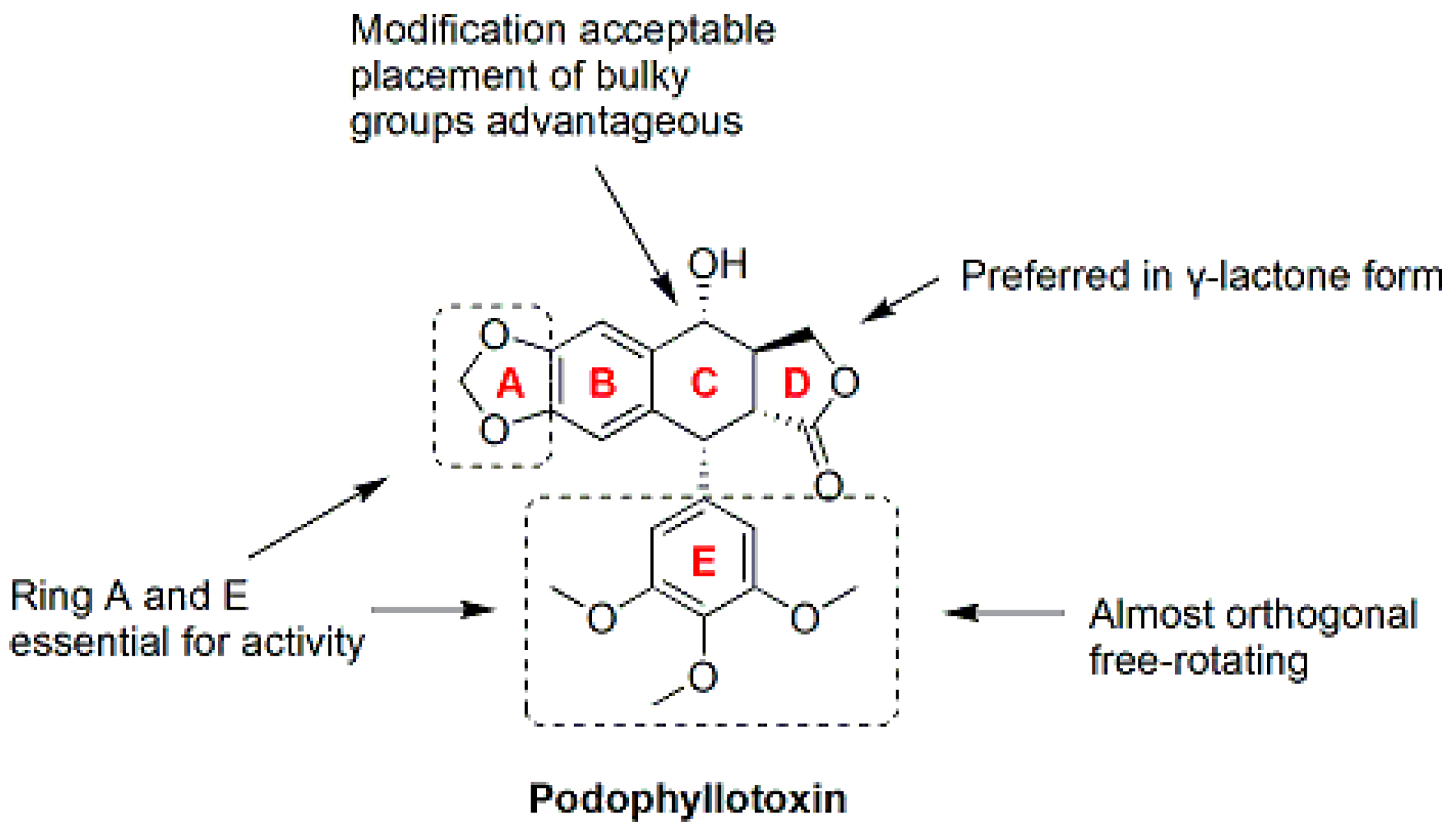
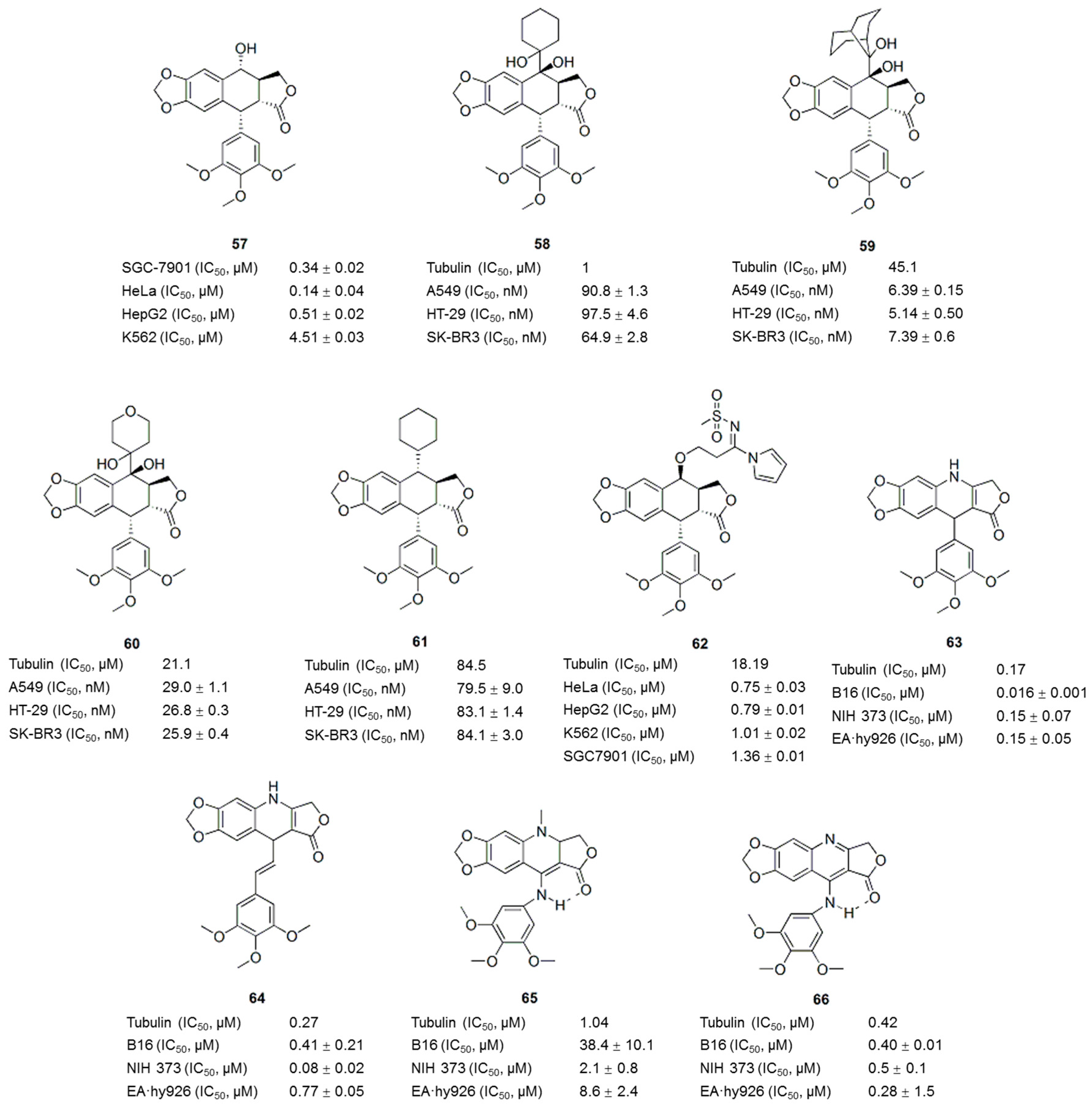
6. Podophyllotoxin Derivatives/Analogues
7. Myoseverin Derivatives
8. Sulfonamides
9. Thiazolidinone, Thiazole and Imidazole Analogues
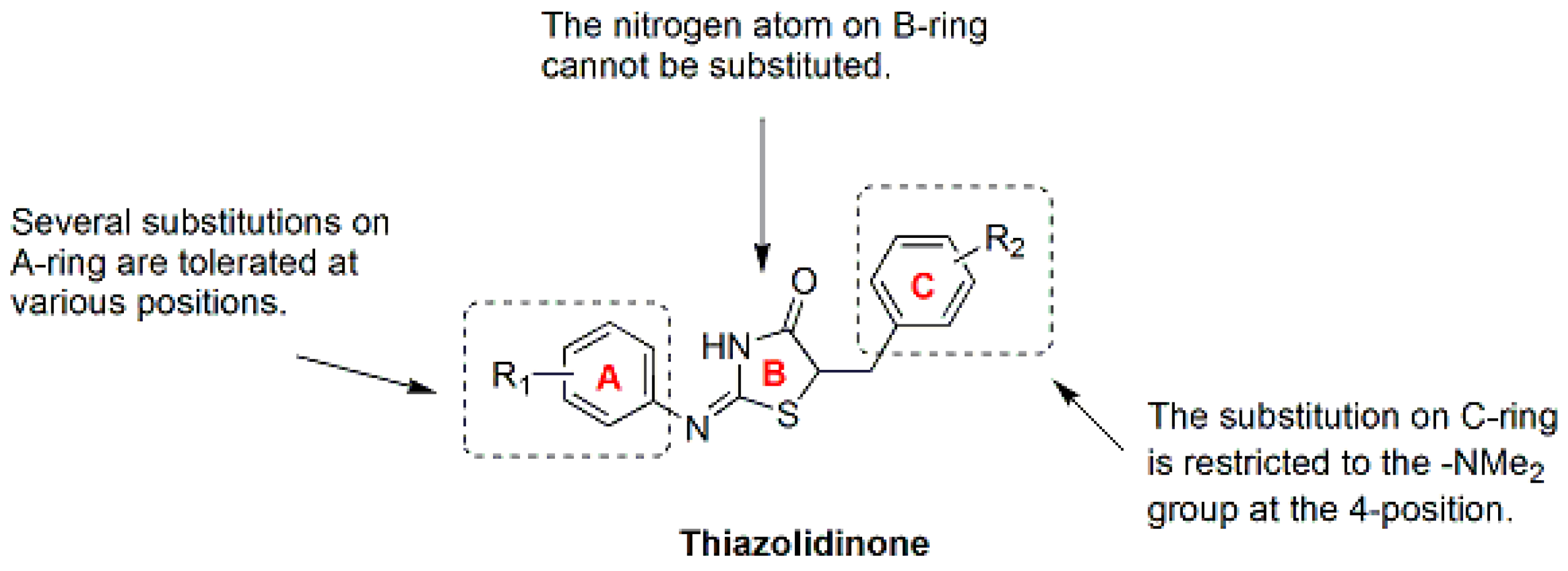
9.1. Thiazolidinone Analogues
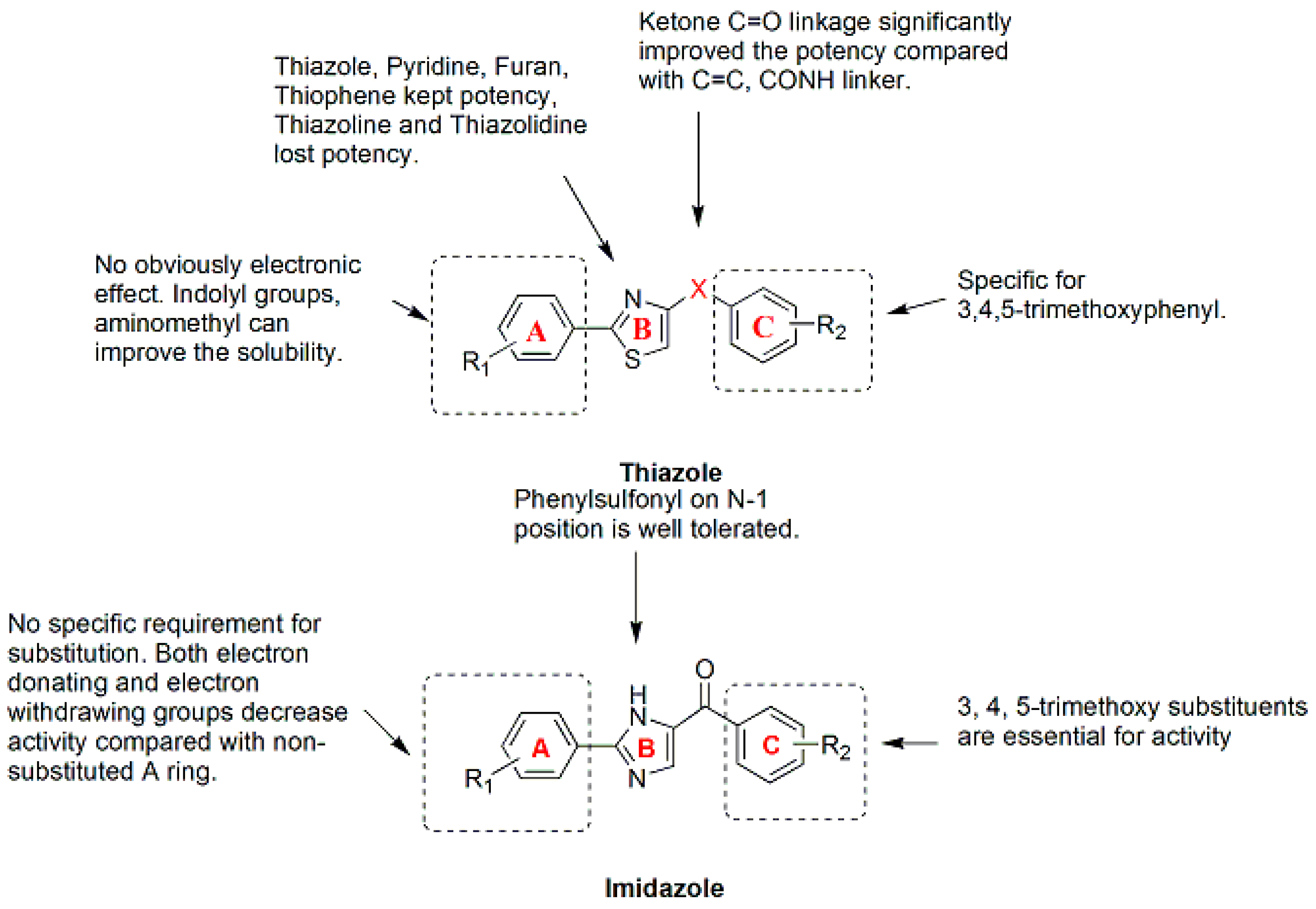
9.2. Thiazole and Imidazols Analogues
10. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wilson, L.; Jordan, M.A. Microtubule dynamics: Taking aim at a moving target. Chem. Biol. 1995, 2, 569–573. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T.; Menefee, M. Mechanisms of multidrug resistance: The potential role of microtubule-stabilizing agents. Ann. Oncol. 2007, 18, v3–v8. [Google Scholar] [CrossRef] [PubMed]
- Buey, R.M.; Barasoain, I.; Jackson, E.; Meyer, A.; Giannakakou, P.; Paterson, I.; Mooberry, S.; Andreu, J.M.; Díaz, J.F. Microtubule interactions with chemically diverse stabilizing agents: Thermodynamics of binding to the paclitaxel site predicts cytotoxicity. Chem. Biol. 2005, 12, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Thrower, D.; Wilson, L. Mechanism of inhibition of cell proliferation by vinca alkaloids. Cancer Res. 1991, 51, 2212–2222. [Google Scholar] [PubMed]
- Jordan, M.A.; Wendell, K.; Gardiner, S.; Derry, W.B.; Copp, H.; Wilson, L. Mitotic block induced in Hela cells by low concentrations of paclitaxel (taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996, 56, 816–825. [Google Scholar] [PubMed]
- Brancale, A.; Silvestri, R. Indole, a core nucleus for potent inhibitors of tubulin polymerization. Med. Res. Rev. 2007, 27, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Flynn, B.L.; Hamel, E.; Jung, M.K. One-pot synthesis of benzo (b) furan and indole inhibitors of tubulin polymerization. J. Med. Chem. 2002, 45, 2670–2673. [Google Scholar] [CrossRef] [PubMed]
- Flynn, B.L.; Gill, G.S.; Grobelny, D.W.; Chaplin, J.H.; Paul, D.; Leske, A.F.; Lavranos, T.C.; Chalmers, D.K.; Charman, S.A.; Kostewicz, E.; et al. Discovery of 7-hydroxy-6-methoxy-2-methyl-3-(3,4,5-trimethoxybenzoyl)benzo (b) furan (BNC105), a tubulin polymerization inhibitor with potent antiproliferative and tumor vascular disrupting properties. J. Med. Chem. 2011, 54, 6014–6027. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, K.; Hatanaka, T.; Fujita, K.; Nakagawa, R.; Fukuda, Y.; Nihei, Y.; Suga, Y.; Morinaga, Y.; Akiyama, Y.; Tsuji, T. Syntheses and antitumor activity of cis-restricted combretastatins: 5-Membered heterocyclic analogues. Bioorg. Med. Chem. Lett. 1998, 8, 3153–3158. [Google Scholar] [CrossRef]
- Banimustafa, M.; Kheirollahi, A.; Safavi, M.; Ardestani, S.K.; Aryapour, H.; Foroumadi, A.; Emami, S. Synthesis and biological evaluation of 3-(trimethoxyphenyl)-2(3H)-thiazole thiones as combretastatin analogs. Eur. J. Med. Chem. 2013, 70, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Woods, K.W.; Li, Q.; Barr, K.J.; McCroskey, R.W.; Hannick, S.M.; Gherke, L.; Credo, R.B.; Hui, Y.H.; Marsh, K.; et al. Potent, orally active heterocycle-based combretastatin A-4 analogues: Synthesis, structure-activity relationship, pharmacokinetics, and in vivo antitumor activity evaluation. J. Med. Chem. 2002, 45, 1697–1711. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Wang, Z.; Li, C.M.; Lu, Y.; Vaddady, P.K.; Meibohm, B.; Dalton, J.T.; Miller, D.D.; Li, W. Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicines binding site in tubulin as potential anticancer agents. J. Med. Chem. 2010, 53, 7414–7427. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, V.; Venghateri, J.B.; Dhaked, H.P.S.; Bhoyar, A.S.; Guchhait, S.K.; Panda, D. Novel combretastatin-2-aminoimidazole analogues as potent tubulin assembly inhibitors: Exploration of unique pharmacophoric impact of bridging skeleton and aryl moiety. J. Med. Chem. 2016, 59, 3439–3451. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, Y.; Aks, S.E.; Hutson, J.R.; Juurlink, D.N.; Nguyen, P.; Dubnov-Raz, G.; Pollak, U.; Koren, G.; Bentur, Y. Colchicine poisoning: The dark side of an ancient drug. Clin. Toxicol. 2010, 48, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Quinn, F.R.; Neiman, Z.; Beisler, J.A. Toxicity quantitative structure-activity relationships of colchicines. J. Med. Chem. 1981, 24, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Ringel, I.; Jaffe, D.; Alerhand, S.; Boye, O.; Muzaffar, A.; Brossi, A. Fluorinated colchicinoids: Antitubulin and cytotoxic properties. J. Med. Chem. 1991, 34, 3334–3338. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Datta, A.B.; Gupta, S.; Poddar, A.; Sengupta, S.; Janik, M.E.; Bhattacharyya, B. -NH-dansyl isocolchicine exhibits a significantly improved tubulin-binding affinity and microtubule inhibition in comparison to isocolchicine by binding tubulin through its A and B rings. Biochemistry 2005, 44, 3249–3258. [Google Scholar] [CrossRef] [PubMed]
- Sapra, S.; Bhalla, Y.; Nandani Sharma, S.; Singh, G.; Nepali, K.; Budhiraja, A.; Dhar, K.L. Colchicine and its various physicochemical and biological aspects. Med. Chem. Res. 2013, 22, 531–547. [Google Scholar] [CrossRef]
- Tang-Wai, D.F.; Brossi, A.; Arnold, L.D.; Gros, P. The nitrogen of the acetamido group of colchicine modulates P-glycoprotein-mediated multidrug resistance. Biochemistry 1993, 32, 6470–6476. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, M.; Schilling, B.; Ravindra, R.; Winter, J.; Janik, M.E. Synthesis and biological evaluation of B-ring modified colchicine and isocolchicine analogs. Bioorgan. Med. Chem. Lett. 2006, 16, 2761–2764. [Google Scholar] [CrossRef] [PubMed]
- Thomopoulou, P.; Sachs, J.; Teusch, N.; Mariappan, A.; Gopalakrishnan, J.; Schmalz, H.-G.N. New colchicine-derived triazoles and their influence on cytotoxicity and microtubule morphology. ACS Med. Chem. Lett. 2015, 7, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, L.; Redondo-Horcajo, M.; Zhao, Y.; Santos, A.R.; Chowdury, K.F.; Vinader, V.; Abdallah, Q.M.A.; Abdel-Rahman, H.; Fournier-Dit-Chabert, J.; Shnyder, S.D.; et al. Synthesis and biological evaluation of colchicine B-ring analogues tethered with halogenated benzyl moieties. J. Med. Chem. 2012, 55, 11062–11066. [Google Scholar] [CrossRef] [PubMed]
- Fournier-Dit-Chabert, J.; Vinader, V.; Santos, A.R.; Redondo-Horcajo, M.; Dreneau, A.; Basak, R.; Cosentino, L.; Marston, G.; Abdel-Rahman, H.; Loadman, P.M.; et al. Synthesis and biological evaluation of colchicine C-ring analogues tethered with aliphatic linkers suitable for prodrug derivatisation. Bioorg. Med. Chem. Lett. 2012, 22, 7693–7696. [Google Scholar] [CrossRef] [PubMed]
- Zefirova, O.N.; Nurieva, E.V.; Shishov, D.V.; Baskin, I.I.; Fuchs, F.; Lemcke, H.; Schroder, F.; Weiss, D.G.; Zefirov, N.S.; Kuznetsov, S.A. Synthesis and SAR requirements of adamantane-colchicine conjugates with both microtubule depolymerizing and tubulin clustering activities. Bioorg. Med. Chem. 2011, 19, 5529–5538. [Google Scholar] [CrossRef] [PubMed]
- Zefirova, O.N.; Lemcke, H.; Lantow, M.; Nurieva, E.V.; Wobith, B.; Onishchenko, G.E.; Hoenen, A.; Griffiths, G.; Zefirov, N.S.; Kuznetsov, S.A. Unusual tubulin-clustering ability of specifically C7-modified colchicine analogues. ChemBioChem 2013, 14, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Kumar, A.; Joshi, P.; Guru, S.K.; Kumar, S.; Wani, Z.A.; Mahajan, G.; Hussain, A.; Qazi, A.K.; Kumar, A.; et al. Colchicine derivatives with potent anticancer activity and reduced P-glycoprotein induction liability. Org. Biomol. Chem. 2015, 13, 5674–5689. [Google Scholar] [CrossRef] [PubMed]
- Huczynski, A.; Rutkowski, J.; Popiel, K.; Maj, E.; Wietrzyk, J.; Stefanska, J.; Majcher, U.; Bartl, F. Synthesis, antiproliferative and antibacterial evaluation of C-ring modified colchicine analogues. Eur. J. Med. Chem. 2015, 90, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Vilanova, C.; Diaz-Oltra, S.; Murga, J.; Falomir, E.; Carda, M.; Redondo-Horcajo, M.; Diaz, J.F.; Barasoain, I.; Marco, J.A. Design and synthesis of pironetin analogue/colchicine hybrids and study of their cytotoxic activity and mechanisms of interaction with tubulin concepcion. J. Med. Chem. 2014, 57, 10391–10403. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.M.; Singh, S.B.; Chu, P.S.; Dempcy, R.O.; Schmidt, J.M.; Pettit, G.R.; Hamel, E. Interactions of tubulin with potent natural and synthetic analogs of the antimitotic agent combretastatin: A structure-activity study. Mol. Pharmacol. 1988, 34, 200–208. [Google Scholar] [PubMed]
- Nam, N.H. Combretastatin A-4 analogues as antimitotic antitumor agents. Curr. Med. Chem. 2003, 10, 1697–1722. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Davis, R.; Vanderham, J.; Hills, P.; Mackay, H.; Brown, T.; Mooberry, S.L.; Lee, M. 1,2,3,4-tetrahydro-2-thioxopyrimidine analogs of combretastatin-A4. Eur. J. Med. Chem. 2008, 43, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Rhodes, M.R.; Herald, D.L.; Hamel, E.; Schmidt, J.M.; Pettitt, R.K. Antineoplastic agents. 445. Synthesis and evaluation of structural modifications of (Z)- and (E)-combretastatin A-4. J. Med. Chem. 2005, 48, 4087–4099. [Google Scholar] [CrossRef] [PubMed]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, S.; Genazzani, A.A. Medicinal chemistry of combretastatin A4: Present and future directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Alloatti, D.; Giannini, G.; Cabri, W.; Lustrati, I.; Marzi, M.; Ciacci, A.; Gallo, G.; Tinti, M.O.; Marcellini, M.; Riccioni, T.; et al. Synthesis and biological activity of fluorinated combretastatin analogues. J. Med. Chem. 2008, 51, 2708–2721. [Google Scholar] [CrossRef] [PubMed]
- Nagaiah, G.; Remick, S.C. Combretastatin A4 phosphate: A novel vascular disrupting agent. Future Oncol. 2010, 6, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Ng, Q.S.; Mandeville, H.; Goh, V.; Alonzi, R.; Milner, J.; Carnell, D.; Meer, K.; Padhani, A.R.; Saunders, M.I.; Hoskin, P.J. Phase Ib trial of radiotherapy in combination with combretastatin-A4-phosphate in patients with non-small-cell lung cancer, prostate adenocarcinoma, and squamous cell carcinoma of the head and neck. Ann. Oncol. 2012, 23, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Delmonte, A.; Sessa, C. Ave8062: A new combretastatin derivative vascular disrupting agent. Expert Opin. Investig. Drugs 2009, 18, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Lippert, J.W. Vascular disrupting agents. Bioorg. Med. Chem. 2007, 15, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Lippert, J.W. Antineoplastic agents 429. Syntheses of the combretastatin A-1 and combretastatin B-1 prodrugs. Anti-Cancer Drug Des. 2000, 15, 203–216. [Google Scholar]
- Devkota, L.; Lin, C.M.; Strecker, T.E.; Wang, Y.F.; Tidmore, J.K.; Chen, Z.; Guddneppanavar, R.; Jelinek, C.J.; Lopez, R.; Liu, L.; et al. Design, synthesis, and biological evaluation of water-soluble amino acid prodrug conjugates derived from combretastatin, dihydronaphthalene, and benzosuberene-based parent vascular disrupting agents. Bioorg. Med. Chem. 2016, 24, 938–956. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.T.; Man, R.J.; Tang, D.J.; Yao, Y.F.; Tao, X.X.; Yu, C.; Liang, X.Y.; Makawana, J.A.; Zou, M.J.; Wang, Z.C.; et al. Design, synthesis and antitumor activity of novel link-bridge and B-ring modified combretastatin A-4 (CA-4) analogues as potent antitubulin agents. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Gerova, M.S.; Stateva, S.R.; Radonova, E.M.; Kalenderska, R.B.; Rusew, R.I.; Nikolova, R.P.; Chanev, C.D.; Shivachev, B.L.; Apostolova, M.D.; Petrov, O.I. Combretastatin A-4 analogues with benzoxazolone scaffold: Synthesis, structure and biological activity. Eur. J. Med. Chem. 2016, 120, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Do, C.V.; Faouzi, A.; Barette, C.; Farce, A.; Fauvarque, M.O.; Colomb, E.; Catry, L.; Berthier-Vergnes, O.; Haftek, M.; Barret, R.; et al. Synthesis and biological evaluation of thiophene and benzo b thiophene analogs of combretastatin A-4 and isocombretastatin A-4: A comparison between the linkage positions of the 3,4,5-trimethoxystyrene unit. Bioorg. Med. Chem. Lett. 2016, 26, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Zuo, D.Y.; Jiang, N.; Qi, H.; Zhai, Y.P.; Bai, Z.S.; Feng, D.J.; Yang, L.; Jiang, M.Y.; Bao, K.; et al. Microwave-assisted synthesis and biological evaluation of 3,4-diaryl maleic anhydride/n-substituted maleimide derivatives as combretastatin A-4 analogues. Bioorg. Med. Chem. Lett. 2015, 25, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Pati, H.N.; Wicks, M.; Holt, H.L.; LeBlanc, R.; Weisbruch, P.; Forrest, L.; Lee, M. Synthesis and biological evaluation of cis-combretastatin analogs and their novel 1,2,3-triazole derivatives. Heterocycl. Commun. 2005, 11, 117–120. [Google Scholar] [CrossRef]
- Odlo, K.; Hentzen, J.; dit Chabert, J.F.; Ducki, S.; Gani, O.; Sylte, I.; Skrede, M.; Florenes, V.A.; Hansen, T.V. 1,5-disubstituted 1,2,3-triazoles as cis-restricted analogues of combretastatin A-4: Synthesis, molecular modeling and evaluation as cytotoxic agents and inhibitors of tubulin. Bioorg. Med. Chem. 2008, 16, 4829–4838. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Brancale, A.; Fu, X.H.; Li, J.; Zhang, S.Z.; Hamel, E.; et al. Synthesis and evaluation of 1,5-disubstituted tetrazoles as rigid analogues of combretastatin A-4 with potent antiproliferative and antitumor activity. J. Med. Chem. 2012, 55, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.; Greene, L.M.; Knox, A.J.S.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Lead identification of conformationally restricted β-lactam type combretastatin analogues: Synthesis, antiproliferative activity and tubulin targeting effects. Eur. J. Med. Chem. 2010, 45, 5752–5766. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Yang, F.S.; Guo, D.D.; Xu, J.W.; Jiang, M.Y.; Liu, C.J.; Bao, K.; Wu, Y.L.; Zhang, W.G. Synthesis and biological evaluation of novel 3,4-diaryl-1,2,5-selenadiazol analogues of combretastatin A-4. Eur. J. Med. Chem. 2014, 87. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, Q.K.; Bai, Z.S.; Sun, J.; Jiang, X.W.; Song, H.R.; Wu, Y.L.; Zhang, W.G. Synthesis and biological evaluation of 2,3-diarylthiophene analogues of combretastatin A-4. MedChemComm 2015, 6, 971–976. [Google Scholar] [CrossRef]
- Sanghai, N.; Jain, V.; Preet, R.; Kandekar, S.; Das, S.; Trivedi, N.; Mohapatra, P.; Priyadarshani, G.; Kashyap, M.; Das, D.; et al. Combretastatin A-4 inspired novel 2-aryl-3-arylamino-imidazo-pyridines/pyrazines as tubulin polymerization inhibitors, antimitotic and anticancer agents. MedChemComm 2014, 5, 766–782. [Google Scholar] [CrossRef]
- Hu, J.H.; Yan, J.; Chen, J.; Pang, Y.Q.; Huang, L.; Li, X.S. Synthesis, biological evaluation and mechanism study of a class of cyclic combretastatin A-4 analogues as novel antitumour agents. MedChemComm 2015, 6, 1318–1327. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Sheng, C.; Lv, Z.; Dong, G.; Wang, T.; Liu, J.; Zhang, M.; Li, L.; Zhang, T.; et al. Design and synthesis of cyclopropylamide analogues of combretastatin-A4 as novel microtubule-stabilizing agents. J. Med. Chem. 2013, 56, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.L.; Zhong, Q.; Mottamal, M.; Zhang, Q.; Zhang, C.D.; LeMelle, E.; McFerrin, H.; Wang, G.D. Design, synthesis, and biological evaluation of novel pyridine-bridged analogues of combretastatin-A4 as anticancer agents. J. Med. Chem. 2014, 57, 3369–3381. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical importance of indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef] [PubMed]
- Alves, F.R.D.; Barreiro, E.J.; Fraga, C.A.M. From nature to drug discovery: The indole scaffold as a “privileged structure”. Mini-Rev. Med. Chem. 2009, 9, 782–793. [Google Scholar] [CrossRef]
- Patil, S.A.; Patil, R.; Miller, D.D. Indole molecules as inhibitors of tubulin polymerization: Potential new anticancer agents. Future Med. Chem. 2012, 4, 2085–2115. [Google Scholar] [CrossRef] [PubMed]
- Giansanti, V.; Piscitelli, F.; Camboni, T.; Prosperi, E.; La Regina, G.; Parks, M.; Silvestri, R.; Scovassi, A.I. Arylthioindoles: Promising compounds against cancer cell proliferation. Oncol. Lett. 2010, 1, 109–112. [Google Scholar] [PubMed]
- De Martino, G.; La Regina, G.; Coluccia, A.; Edler, M.C.; Barbera, M.C.; Brancale, A.; Wilcox, E.; Hamel, E.; Artico, M.; Silvestri, R. Arylthioindoles, potent inhibitors of tubulin polymerization. J. Med. Chem. 2004, 47, 6120–6123. [Google Scholar] [CrossRef] [PubMed]
- De Martino, G.; Edler, M.C.; La Regina, G.; Coluccia, A.; Barbera, M.C.; Barrow, D.; Nicholson, R.I.; Chiosis, G.; Brancale, A.; Hamel, E.; et al. New arylthioindoles: Potent inhibitors of tubulin polymerization. 2. Structure-activity relationships and molecular modeling studies. J. Med. Chem. 2006, 49, 947–954. [Google Scholar] [CrossRef] [PubMed]
- La Regina, G.; Edler, M.C.; Brancale, A.; Kandil, S.; Coluccia, A.; Piscitelli, F.; Hamel, E.; De Martino, G.; Matesanz, R.; Diaz, J.F.; et al. Arylthioindole inhibitors of tubulin polymerization. 3. Biological evaluation, structure-activity relationships and molecular modeling studies. J. Med. Chem. 2007, 50, 2865–2874. [Google Scholar] [CrossRef] [PubMed]
- La Regina, G.; Sarkar, T.; Bai, R.L.; Edler, M.C.; Saletti, R.; Coluccia, A.; Piscitelli, F.; Minelli, L.; Gatti, V.; Mazzoccoli, C.; et al. New arylthioindoles and related bioisosteres at the sulfur bridging group. 4. Synthesis, tubulin polymerization, cell growth inhibition, and molecular modeling studies. J. Med. Chem. 2009, 52, 7512–7527. [Google Scholar] [CrossRef] [PubMed]
- La Regina, G.; Bai, R.; Rensen, W.; Coluccia, A.; Piscitelli, F.; Gatti, V.; Bolognesi, A.; Lavecchia, A.; Granata, I.; Porta, A.; et al. Design and synthesis of 2-heterocyclyl-3-arylthio-1H-indoles as potent tubulin polymerization and cell growth inhibitors with improved metabolic stability. J. Med. Chem. 2011, 54, 8394–8406. [Google Scholar] [CrossRef] [PubMed]
- La Regina, G.; Bai, R.L.; Rensen, W.M.; Di Cesare, E.; Coluccia, A.; Piscitelli, F.; Famiglini, V.; Reggio, A.; Nalli, M.; Pelliccia, S.; et al. Toward highly potent cancer agents by modulating the C-2 group of the arylthioindole class of tubulin polymerization inhibitors. J. Med. Chem. 2013, 56, 123–149. [Google Scholar] [CrossRef] [PubMed]
- Colley, H.E.; Muthana, M.; Danson, S.J.; Jackson, L.V.; Brett, M.L.; Harrison, J.; Coole, S.F.; Mason, D.P.; Jennings, L.R.; Wong, M.; et al. An orally bioavailable, indole-3-glyoxylamide based series of tubulin polymerization inhibitors showing tumor growth inhibition in a mouse xenograft model of head and neck cancer. J. Med. Chem. 2015, 58, 9309–9333. [Google Scholar] [CrossRef] [PubMed]
- MacDonough, M.T.; Strecker, T.E.; Hamel, E.; Hall, J.J.; Chaplin, D.J.; Trawick, M.L.; Pinney, K.G. Synthesis and biological evaluation of indole-based, anti-cancer agents inspired by the vascular disrupting agent 2-(3′-hydroxy-4′-methoxyphenyl)-3-(3″,4″,5″-trimethoxybenzoyl)-6-methoxyindole (oxi8006). Bioorgan. Med. Chem. 2013, 21, 6831–6843. [Google Scholar] [CrossRef] [PubMed]
- La Regina, G.; Bai, R.L.; Coluccia, A.; Farniglini, V.; Pelliccia, S.; Passacantilli, S.; Mazzoccoli, C.; Ruggieri, V.; Verrico, A.; Miele, A.; et al. New indole tubulin assembly inhibitors cause stable arrest of mitotic progression, enhanced stimulation of natural killer cell cytotoxic activity, and repression of hedgehog-dependent cancer. J. Med. Chem. 2015, 58, 5789–5807. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.Y.; Xu, J.W.; Wang, Z.W.; Qi, H.; Xu, Q.L.; Bai, Z.S.; Zhang, Q.; Bao, K.; Wu, Y.L.; Zhang, W.G. 3-(3,4,5-trimethoxyphenylselenyl)-1H-indoles and their selenoxides as combretastatin A-4 analogs: Microwave-assisted synthesis and biological evaluation. Eur. J. Med. Chem. 2015, 90, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Reddy, V.S.; Karnewar, S.; Chourasiya, S.S.; Shaik, A.B.; Kumar, G.B.; Kishor, C.; Reddy, M.K.; Narasimha Rao, M.; Nagabhushana, A. Synthesis and biological evaluation of imidazopyridine-oxindole conjugates as microtubule-targeting agents. ChemMedChem 2013, 8, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.J.; Wang, J.; Li, W.; Miller, D.D. Structural optimization of indole derivatives acting at colchicine binding site as potential anticancer agents. ACS Med. Chem. Lett. 2015, 6, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.J.; Zhang, B.; Yang, H.K.; Liu, Y.; Chen, Y.R.; Ma, T.Z.; Lu, L.; You, W.W.; Zhao, P.L. Design, synthesis and molecular docking studies of novel indole-pyrimidine hybrids as tubulin polymerization inhibitors. Chem. Biol. Drug Des. 2015, 86, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Orlikova, B.; Tasdemir, D.; Golais, F.; Dicato, M.; Diederich, M. Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr. 2011, 6, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.M.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, N.J.; Rennison, D.; McGown, A.T.; Ducki, S.; Gul, L.A.; Hadfield, J.A.; Khan, N. Linked parallel synthesis and MTT bioassay screening of substituted chalcones. J. Comb. Chem. 2001, 3, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Ducki, S.; Rennison, D.; Woo, M.; Kendall, A.; Chabert, J.F.D.; McGown, A.T.; Lawrence, N.J. Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: Synthesis and biological evaluation of antivascular activity. Bioorg. Med. Chem. 2009, 17, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Ma, G.Y.; Yang, Y.; Cheng, K.; Zheng, Q.Z.; Mao, W.J.; Shi, L.; Zhao, J.; Zhu, H.L. Synthesis, molecular modeling and biological evaluation of dithiocarbamates as novel antitubulin agents. Bioorg. Med. Chem. 2010, 18, 4310–4316. [Google Scholar] [CrossRef] [PubMed]
- Salum, L.B.; Altei, W.F.; Chiaradia, L.D.; Cordeiro, M.N.S.; Canevarolo, R.R.; Melo, C.P.S.; Winter, E.; Mattei, B.; Daghestani, H.N.; Santos-Silva, M.C.; et al. Cytotoxic 3,4,5-trimethoxychalcones as mitotic arresters and cell migration inhibitors. Eur. J. Med. Chem. 2013, 63, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Qin, Y.J.; Zhang, Y.L.; Li, Y.J.; Rao, B.; Zhang, Y.Q.; Yang, M.R.; Jiang, A.Q.; Qi, J.L.; Zhu, H.L. Synthesis, biological evaluation, and molecular docking studies of novel chalcone oxime derivatives as potential tubulin polymerization inhibitors. RSC Adv. 2014, 4, 32263–32275. [Google Scholar]
- Zhu, C.G.; Zuo, Y.L.; Wang, R.M.; Liang, B.X.; Yue, X.; Wen, G.S.; Shang, N.N.; Huang, L.; Chen, Y.; Du, J.; et al. Discovery of potent cytotoxic ortho-aryl chalcones as new scaffold targeting tubulin and mitosis with affinity-based fluorescence. J. Med. Chem. 2014, 57, 6364–6382. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.C.; Li, C.Y.; He, L.; Lei, K.; Wang, F.; Pu, Y.Z.; Yang, Z.; Cao, D.; Ma, L.; Chen, J.Y.; et al. Design, synthesis and biological evaluation of a series of pyrano chalcone derivatives containing indole moiety as novel anti-tubulin agents. Bioorg. Med. Chem. 2014, 22, 2060–2079. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yan, J.; Hu, J.H.; Pang, Y.Q.; Huang, L.; Li, X.S. Synthesis, biological evaluation and mechanism study of chalcone analogues as novel anti-cancer agents. RSC Adv. 2015, 5, 68128–68135. [Google Scholar] [CrossRef]
- Yan, J.; Chen, J.; Zhang, S.; Hu, J.H.; Huang, L.; Li, X.S. Synthesis, evaluation, and mechanism study of novel indole-chalcone derivatives exerting effective antitumor activity through microtubule destabilization in vitro and in vivo. J. Med. Chem. 2016, 59, 5264–5283. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Kaur, M.; Holzer, W. Synthesis and evaluation of indole, pyrazole, chromone and pyrimidine based conjugates for tumor growth inhibitory activities—Development of highly efficacious cytotoxic agents. Eur. J. Med. Chem. 2010, 45, 4968–4982. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Balakrishna, M.; Nayak, V.L.; Shaik, T.B.; Faazil, S.; Nimbarte, V.D. Design and synthesis of imidazo 2,1-B thiazole-chalcone conjugates: Microtubule-destabilizing agents. Mini-Rev. Med. Chem. 2014, 9, 2766–2780. [Google Scholar] [CrossRef] [PubMed]
- Ruan, B.F.; Lu, X.A.; Tang, J.F.; Wei, Y.; Wang, X.L.; Zhang, Y.B.; Wang, L.S.; Zhu, H.L. Synthesis, biological evaluation, and molecular docking studies of resveratrol derivatives possessing chalcone moiety as potential antitubulin agents. Bioorg. Med. Chem. 2011, 19, 2688–2695. [Google Scholar] [CrossRef] [PubMed]
- Imbert, T.F. Discovery of podophyllotoxins. Biochimie 1998, 80, 207–222. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Tian, J.; Qian, K.; Zhao, X.B.; Morris-Natschke, S.L.; Yang, L.; Nan, X.; Tian, X.; Lee, K.H. Recent progress on C-4-modified podophyllotoxin analogs as potent antitumor agents. Med. Res. Rev. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M.; Castro, M.A.; del Corral, J.M.M.; San Feliciano, A. Antitumor properties of podophyllotoxin and related compounds. Curr. Pharm. Des. 2000, 6, 1811–1839. [Google Scholar] [CrossRef] [PubMed]
- You, Y.J. Podophyllotoxin derivatives: Current synthetic approaches for new anticancer agents. Curr. Pharm. Des. 2005, 11, 1695–1717. [Google Scholar] [CrossRef] [PubMed]
- Abad, A.; Lopez-Perez, J.L.; del Olmo, E.; Garcia-Fernandez, L.F.; Francesch, A.; Trigili, C.; Barasoain, I.; Andreu, J.M.; Diaz, J.F.; San Feliciano, A. Synthesis and antimitotic and tubulin interaction profiles of novel pinacol derivatives of podophyllotoxins. J. Med. Chem. 2012, 55, 6724–6737. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Ojha, R.; Sharma, S.; Bedi, P.M.S.; Dhar, K.L. Tubulin inhibitors: A patent survey. Recent Pat. Anti-Cancer Drug Discov. 2014, 9, 176–220. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Wei, D.F.; Zhao, Y.L.; Cheng, W.D.; Lu, Y.; Ma, Y.Q.; Li, X.; Han, C.; Wei, Y.X.; Cao, H.M.; et al. Synthesis and biological evaluation of a series of podophyllotoxins derivatives as a class of potent antitubulin agents. Bioorg. Med. Chem. 2012, 20, 6285–6295. [Google Scholar] [CrossRef] [PubMed]
- Labruere, R.; Gautier, B.; Testud, M.; Seguin, J.; Lenoir, C.; Desbene-Finck, S.; Helissey, P.; Garbay, C.; Chabot, G.G.; Vidal, M.; et al. Design, synthesis, and biological evaluation of the first podophyllotoxin analogues as potential vascular-disrupting agents. Mini-Rev. Med. Chem. 2010, 5, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Rosania, G.R.; Chang, Y.-T.; Perez, O.; Sutherlin, D.; Dong, H.; Lockhart, D.J.; Schultz, P.G. Myoseverin, a microtubule-binding molecule with novel cellular effects. Nat. Biotechnol. 2000, 18, 304–308. [Google Scholar] [PubMed]
- Moon, H.-S.; Jacobson, E.M.; Khersonsky, S.M.; Luzung, M.R.; Walsh, D.P.; Xiong, W.; Lee, J.W.; Parikh, P.B.; Lam, J.C.; Kang, T.-W.; et al. A novel microtubule destabilizing entity from orthogonal synthesis of triazine library and zebrafish embryo screening. J. Am. Chem. Soc. 2002, 124, 11608–11609. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-T.; Wignall, S.M.; Rosania, G.R.; Gray, N.S.; Hanson, S.R.; Su, A.I.; Merlie, J.; Moon, H.-S.; Sangankar, S.B.; Perez, O. Synthesis and biological evaluation of myoseverin derivatives: Microtubule assembly inhibitors. J. Med. Chem. 2001, 44, 4497–4500. [Google Scholar] [CrossRef] [PubMed]
- Krystof, V.; Moravcova, D.; Paprskarova, M.; Barbier, P.; Peyrot, V.; Hlobilkova, A.; Havlicek, L.; Strnad, M. Synthesis and biological activity of 8-azapurine and pyrazolo 4,3-d pyrimidine analogues of myoseverin. Eur. J. Med. Chem. 2006, 41, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Perez, O.D.; Chang, Y.-T.; Rosania, G.; Sutherlin, D.; Schultz, P.G. Inhibition and reversal of myogenic differentiation by purine-based microtubule assembly inhibitors. Chem. Biol. 2002, 9, 475–483. [Google Scholar] [CrossRef]
- Popowycz, F.; Schneider, C.; DeBonis, S.; Skoufias, D.A.; Kozielski, F.; Galmarini, C.M.; Joseph, B. Synthesis and antiproliferative evaluation of pyrazolo[1,5-a]-1,3,5-triazine myoseverin derivatives. Bioorgan. Med. Chem. 2009, 17, 3471–3478. [Google Scholar] [CrossRef] [PubMed]
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C.T. Anticancer and antiviral sulfonamides. Curr. Med. Chem. 2003, 10, 925–953. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Poddar, A.; Mitra, G.; Surolia, A.; Owa, T.; Bhattacharyya, B. Sulfonamide drugs binding to the colchicine site of tubulin: Thermodynamic analysis of the drug-tubulin interactions by isothermal titration calorimetry. J. Med. Chem. 2005, 48, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.X.; Li, Z.R.; Jiang, J.D.; Boykin, D.W. Novel diaryl or heterocyclic sulfonamides as antimitotic agents. Anti-Cancer Agents Med. Chem. 2008, 8, 739–745. [Google Scholar] [CrossRef]
- Yoshino, H.; Ueda, N.; Niijima, J.; Sugumi, H.; Kotake, Y.; Koyanagi, N.; Yoshimatsu, K.; Asada, M.; Watanabe, T.; Nagasu, T. Novel sulfonamides as potential, systemically active antitumor agents. J. Med. Chem. 1992, 35, 2496–2497. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Noda, K.; Yoshimura, A.; Fukuoka, M.; Furuse, K.; Niitani, H. Phase I study of E7010. Cancer Chemother. Pharmacol. 1998, 42, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Hande, K.R.; Hagey, A.; Berlin, J.; Cai, Y.N.; Meek, K.; Kobayashi, H.; Lockhart, A.C.; Medina, D.; Sosman, J.; Gordon, G.B.; et al. The pharmacokinetics and safety of ABT-751, a novel, orally bioavailable sulfonamide antimitotic agent: Results of a phase 1 study. Clin. Cancer Res. 2006, 12, 2834–2840. [Google Scholar] [CrossRef] [PubMed]
- Mauer, A.M.; Cohen, E.E.W.; Ma, P.C.; Kozloff, M.F.; Schwartzberg, L.; Coates, A.I.; Qian, J.; Hagey, A.E.; Gordon, G.B. A phase ii study of ABT-751 in patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 2008, 3, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Owa, T.; Okauchi, T.; Yoshimatsu, K.; Sugi, N.H.; Ozawa, Y.; Nagasu, T.; Koyanagi, N.; Okabe, T.; Kitoh, K.; Yoshino, H. A focused compound library of novel n-(7-indolyl)benzenesulfonamides for the discovery of potent cell cycle inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 1223–1226. [Google Scholar] [CrossRef]
- Yokoi, A.; Kuromitsu, J.; Kawai, T.; Nagasu, T.; Sugi, N.H.; Yoshimatsu, K.; Yoshino, H.; Owa, T. Profiling novel sulfonamide antitumor agents with cell-based phenotypic screens and array-based gene expression analysis. Mol. Cancer Ther. 2002, 1, 275–286. [Google Scholar] [PubMed]
- Chang, J.Y.; Hsieh, H.P.; Chang, C.Y.; Hsu, K.S.; Chiang, Y.F.; Chen, C.M.; Kuo, C.C.; Liou, J.P. 7-aroyl-aminoindoline-1-sulfonamides as a novel class of potent antitubulin agents. J. Med. Chem. 2006, 49, 6656–6659. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.X.; Li, Z.R.; Wang, Y.M.; Wu, Y.B.; Jiang, J.D.; Boykin, D.W. Novel pyridinyl and pyrimidinylcarbazole sulfonamides as antiproliferative agents. Bioorg. Med. Chem. Lett. 2007, 17, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Hu, L.X.; Liu, Z.M.; You, X.F.; Zhang, S.H.; Qu, J.R.; Li, Z.R.; Li, Y.; Kong, W.J.; He, H.W.; et al. N-(2,6-dimethoxypyridine-3-yl)-9-methylcarbazole-3-sulfonamide as a novel tubulin ligand against human cancer. Clin. Cancer Res. 2008, 14, 6218–6227. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.C.; Shan, B.; Beckmann, H.; Farrell, R.P.; Clark, D.L.; Learned, R.M.; Roche, D.; Li, A.; Baichwal, V.; Case, C.; et al. Novel antineoplastic agents with efficacy against multidrug resistant tumor cells. Bioorg. Med. Chem. Lett. 1998, 8, 2653–2656. [Google Scholar] [CrossRef]
- Shan, B.; Medina, J.C.; Santha, E.; Frankmoelle, W.P.; Chou, T.C.; Learned, R.M.; Narbut, M.R.; Stott, D.; Wu, P.G.; Jaen, J.C.; et al. Selective, covalent modification of β-tubulin residue CYS-239 by t138067, an antitumor agent with in vivo efficacy against multidrug-resistant tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 5686–5691. [Google Scholar] [CrossRef] [PubMed]
- Abbassi, N.; Chicha, H.; Rakib, E.; Hannioui, A.; Alaoui, M.; Hajjaji, A.; Geffken, D.; Aiello, C.; Gangemi, R.; Rosano, C.; et al. Synthesis, antiproliferative and apoptotic activities of n-(6(4)-indazolyl)-benzenesulfonamide derivatives as potential anticancer agents. Eur. J. Med. Chem. 2012, 57, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.L.; Tian, W.; Wang, Y.; Kuang, S.; Luo, X.M.; Yu, Q. A novel sulfonamide agent, MPSP-001, exhibits potent activity against human cancer cells in vitro through disruption of microtubule. Acta Pharmacol. Sin. 2012, 33, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Aceves-Luquero, C.; Galiana-Roselló, C.; Ramis, G.; Villalonga-Planells, R.; García-España, E.; Fernández de Mattos, S.; Peláez, R.; Llinares, J.M.; González-Rosende, M.E.; Villalonga, P. N-(2-methyl-indol-1H-5-yl)-1-naphthalenesulfonamide: A novel reversible antimitotic agent inhibiting cancer cell motility. Biochem. Pharmacol. 2016, 115, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.R.; Mallireddigari, M.R.; Pallela, V.R.; Cosenza, S.C.; Billa, V.K.; Akula, B.; Subbaiah, D.; Bharathi, E.V.; Padgaonkar, A.; Lv, H.; et al. Design, synthesis, and biological evaluation of (E)-n-aryl-2-arylethenesulfonamide analogues as potent and orally bioavailable microtubule-targeted anticancer agents. J. Med. Chem. 2013, 56, 5562–5586. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Saraf, S.K. 4-thiazolidinone—A biologically active scaffold. Eur. J. Med. Chem. 2008, 43, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhou, H.Y.; Zhai, S.M.; Yan, B. Natural product-inspired synthesis of thiazolidine and thiazolidinone compounds and their anticancer activities. Curr. Pharm. Des. 2010, 16, 1826–1842. [Google Scholar] [CrossRef] [PubMed]
- Teraishi, F.; Wu, S.; Sasaki, J.; Zhang, L.; Davis, J.; Guo, W.; Dong, F.; Fang, B. Jnk1-dependent antimitotic activity of thiazolidin compounds in human non-small-cell lung and colon cancer cells. Cell. Mol. Life Sci. 2005, 62, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Teraishi, F.; Wu, S.; Sasaki, J.; Zhang, L.; Zhu, H.-B.; Davis, J.J.; Fang, B. P-glycoprotein-independent apoptosis induction by a novel synthetic compound, MMPT [5-[(4-methylphenyl) methylene]-2-(phenylamino)-4(5H)-thiazolone]. J. Pharmacol. Exp. Ther. 2005, 314, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Guo, W.; Teraishi, F.; Pang, J.; Kaluarachchi, K.; Zhang, L.; Davis, J.; Dong, F.; Yan, B.; Fang, B. Anticancer activity of 5-benzylidene-2-phenylimino-1, 3-thiazolidin-4-one (BPT) analogs. Med. Chem. 2006, 2, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Teraishi, F.; Wu, S.; Zhang, L.; Guo, W.; Davis, J.J.; Dong, F.; Fang, B. Identification of a novel synthetic thiazolidin compound capable of inducing c-Jun NH2-terminal Kinase-Dependent apoptosis in human colon cancer cells. Cancer Res. 2005, 65, 6380–6387. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Zhai, S.; Liu, A.; Sun, Y.; Li, R.; Zhang, Y.; Ekins, S.; Swaan, P.W.; Fang, B. Design, synthesis, cytoselective toxicity, structure-activity relationships, and pharmacophore of thiazolidinone derivatives targeting drug-resistant lung cancer cells. J. Med. Chem. 2008, 51, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, Q.; Liu, A.; Li, X.; Zhou, H.; Liu, Y.; Yan, B. Proteome interrogation using nanoprobes to identify targets of a cancer-killing molecule. J. Am. Chem. Soc. 2011, 133, 6886–6889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhai, S.; Li, X.; Zhang, Q.; Wu, L.; Liu, Y.; Jiang, C.; Zhou, H.; Li, F.; Zhang, S. Synergistic action by multi-targeting compounds produces a potent compound combination for human NSCLC both in vitro and in vivo. Cell Death Dis. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhai, S.; Li, L.; Li, X.; Zhou, H.; Liu, A.; Su, G.; Mu, Q.; Du, Y.; Yan, B. Anti-tumor selectivity of a novel tubulin and HSP90 dual-targeting inhibitor in non-small cell lung cancer models. Biochem. Pharmacol. 2013, 86, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhai, S.; Li, L.; Li, X.; Jiang, C.; Zhang, C.; Yan, B. P-glycoprotein-evading anti-tumor activity of a novel tubulin and HSP90 dual inhibitor in a non-small-cell lung cancer model. J. Pharmacol. Sci. 2014, 126, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Liu, Y.; Li, L.; Tian, C.; Zhou, H.; Zhang, Q.; Yan, B. The novel tubulin polymerization inhibitor mhpt exhibits selective anti-tumor activity against rhabdomyosarcoma in vitro and in vivo. PLoS ONE 2015, 10, e0121806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, X.; Li, X.; Li, C.; Zhou, H.; Yan, B. Antitumor activity of (2E,5Z)-5-(2-hydroxybenzylidene)-2-((4-phenoxyphenyl) imino) thiazolidin-4-one, a novel microtubule-depolymerizing agent, in U87MG human glioblastoma cells and corresponding mouse xenograft model. J. Pharmacol. Sci. 2013, 122, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, Y.; Zhang, Q.; Zhou, H.; Zhang, Y.; Yan, B. Comparison of cancer cell survival triggered by microtubule damage after turning DYRK1B kinase on and off. ACS Chem. Biol. 2014, 9, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Choy, M.; Ngo, L.; Foster, S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.C.; Jiang, S.; Zhou, X.C.; Hou, X.; Jin, F.; Podratz, K.C.; Jiang, S.-W. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol. Cancer Ther. 2006, 5, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Zuco, V.; de Cesare, M.; Cincinelli, R.; Nannei, R.; Pisano, C.; Zaffaroni, N.; Zunino, F. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLoS ONE 2011, 6, e29085. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, F.; Wu, L.; Wang, W.; Zhu, H.; Zhang, Q.; Zhou, H.; Yan, B. Improving both aqueous solubility and anti-cancer activity by assessing progressive lead optimization libraries. Bioorg. Med. Chem. Lett. 2015, 25, 1971–1975. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, C.M.; Wang, Z.; Ross, C.R.; Chen, J.; Dalton, J.T.; Li, W.; Miller, D.D. Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as novel anticancer agents: Synthesis, biological evaluation, and structure-activity relationships. J. Med. Chem. 2009, 52, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lu, Y.; Li, W.; Miller, D.D.; Mahato, R.I. Synthesis, formulation and in vitro evaluation of a novel microtubule destabilizer, smart-100. J. Control. Release 2010, 143, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, C.M.; Wang, Z.; Chen, J.J.; Mohler, M.L.; Li, W.; Dalton, J.T.; Miller, D.D. Design, synthesis, and SAR studies of 4-substituted methoxylbenzoyl-aryl-thiazoles analogues as potent and orally bioavailable anticancer agents. J. Med. Chem. 2011, 54, 4678–4693. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.J.; Wang, J.; Li, C.M.; Ahn, S.; Barrett, C.M.; Dalton, J.T.; Li, W.; Miller, D.D. Design, synthesis, and biological evaluation of stable colchicine binding site tubulin inhibitors as potential anticancer agents. J. Med. Chem. 2014, 57, 7355–7366. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, Z.; Liu, Y.B.; Ma, L.; Wu, Y.Z.; He, L.; Shao, M.F.; Yu, K.; Wu, W.S.; Pu, Y.Z.; et al. Synthesis and biological evaluation of diarylthiazole derivatives as antimitotic and antivascular agents with potent antitumor activity. Bioorg. Med. Chem. 2015, 23, 3337–3350. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Ahn, S.J.; Wang, J.; Chen, J.J.; Miller, D.D.; Dalton, J.T.; Li, W. Discovery of 4-aryl-2-benzoyl-imidazoles as tubulin polymerization inhibitor with potent antiproliferative properties. J. Med. Chem. 2013, 56, 3318–3329. [Google Scholar] [CrossRef] [PubMed]
- Assadieskandar, A.; Amini, M.; Ostad, S.N.; Riazi, G.H.; Cheraghi-Shavi, T.; Shafiei, B.; Shafiee, A. Design, synthesis, cytotoxic evaluation and tubulin inhibitory activity of 4-aryl-5-(3,4,5-trimethoxyphenyl)-2-alkylthio-1H-imidazole derivatives. Bioorg. Med. Chem. 2013, 21, 2703–2709. [Google Scholar] [CrossRef] [PubMed]














© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, M.; Liu, F.; Zhou, H.; Zhai, S.; Yan, B. Novel Natural Product- and Privileged Scaffold-Based Tubulin Inhibitors Targeting the Colchicine Binding Site. Molecules 2016, 21, 1375. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21101375
Dong M, Liu F, Zhou H, Zhai S, Yan B. Novel Natural Product- and Privileged Scaffold-Based Tubulin Inhibitors Targeting the Colchicine Binding Site. Molecules. 2016; 21(10):1375. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21101375
Chicago/Turabian StyleDong, Mengqi, Fang Liu, Hongyu Zhou, Shumei Zhai, and Bing Yan. 2016. "Novel Natural Product- and Privileged Scaffold-Based Tubulin Inhibitors Targeting the Colchicine Binding Site" Molecules 21, no. 10: 1375. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21101375