
NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. KP1019 and NAMI-A, Two Structurally Similar Ruthenium Complexes for Cancer Treatment: Introductive Remarks
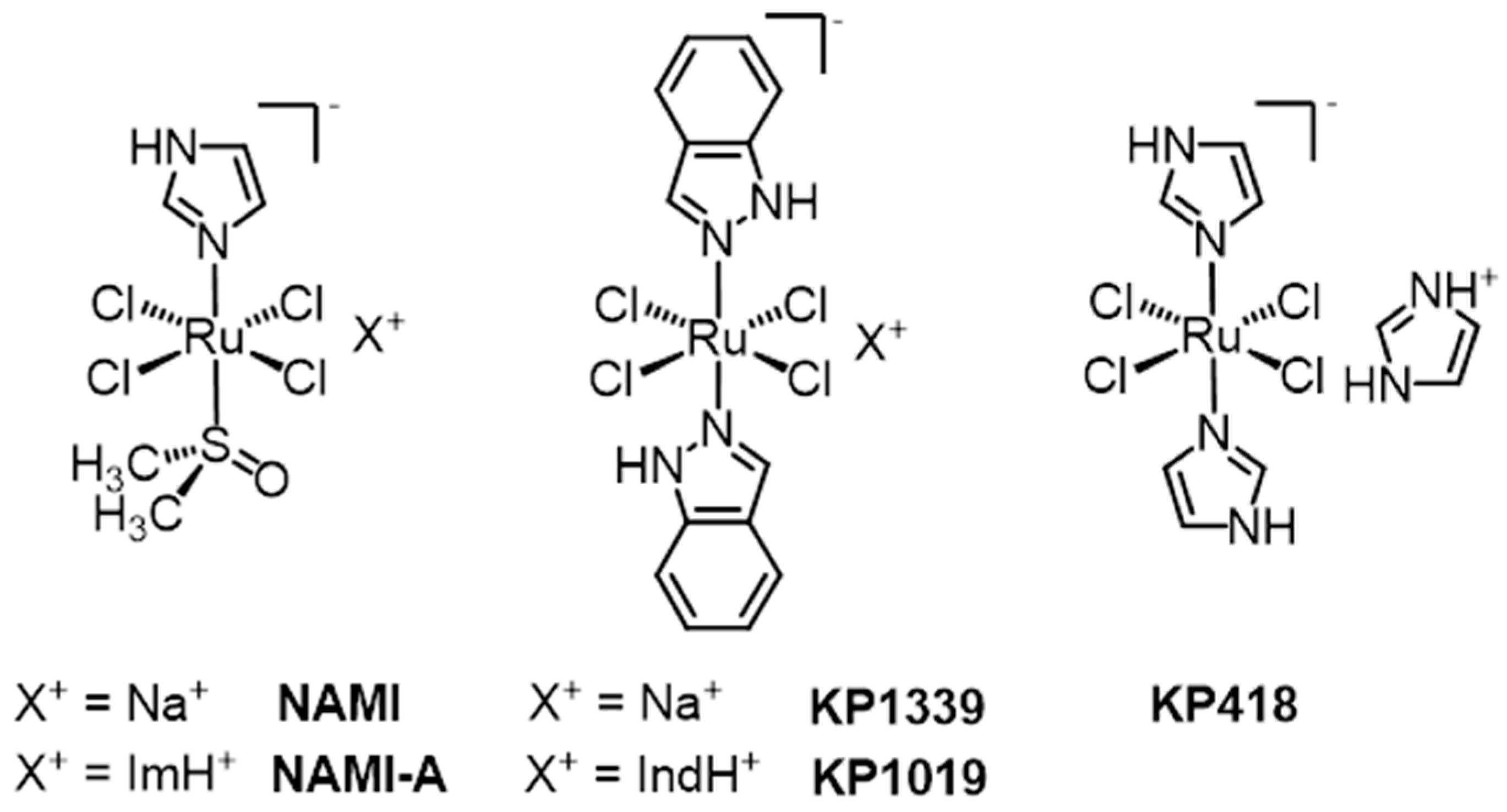
2. KP1019 and NAMI-A: Structural and Solution Chemistry; Biomolecular Interactions
2.1. Structural and Solution Chemistry
2.2. Biomolecular Interactions
2.2.1. Nucleic Acids
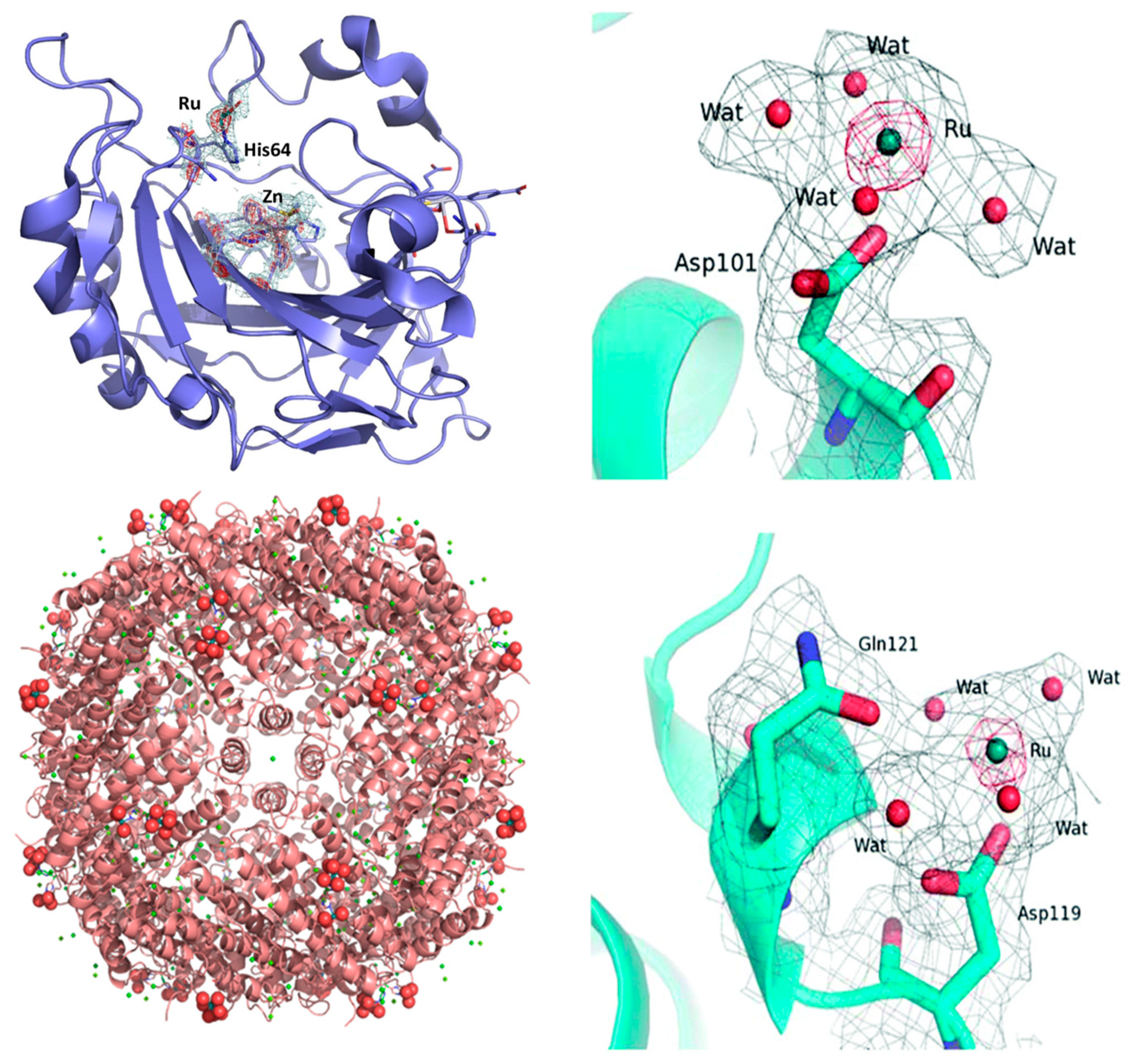
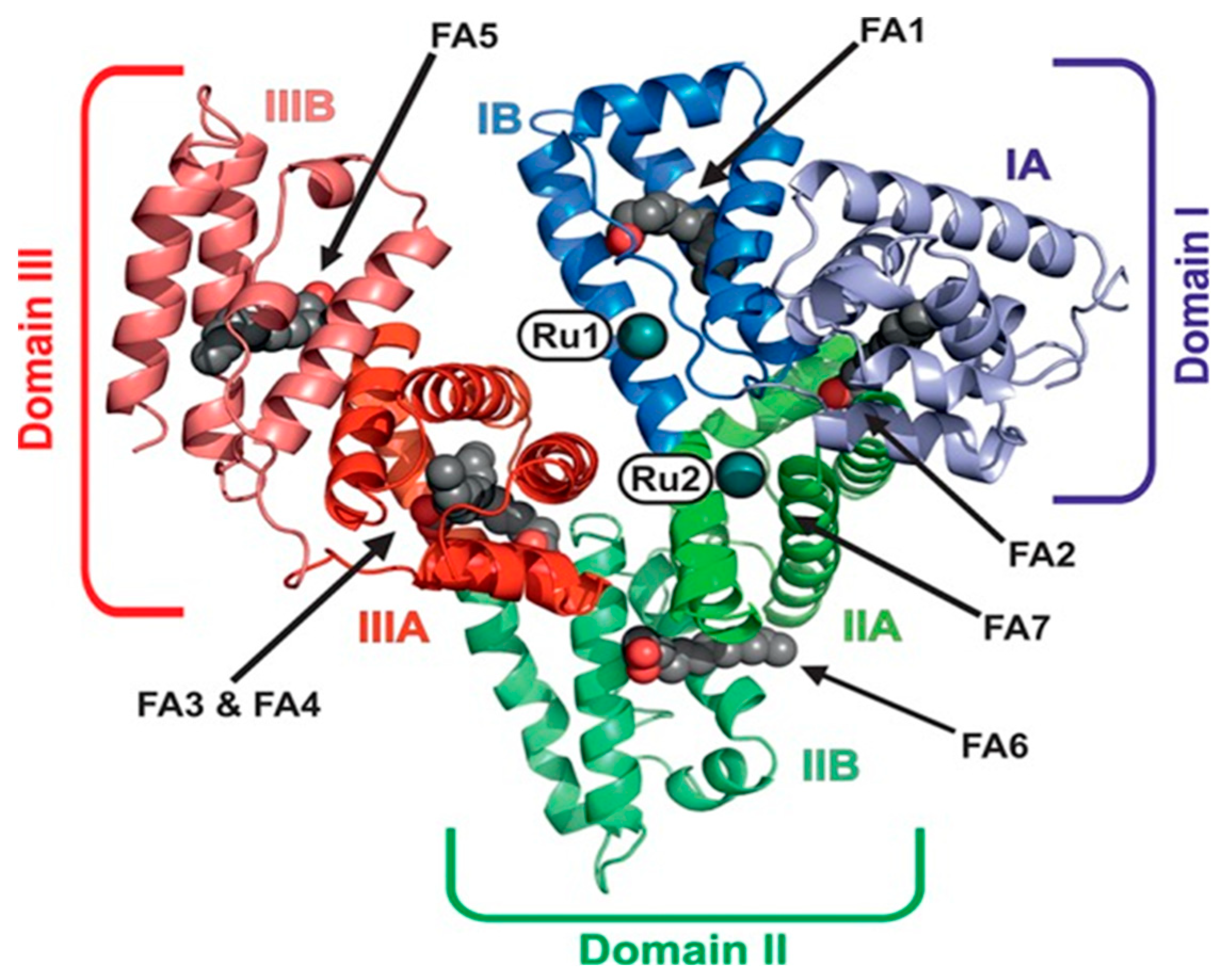
2.2.2. Proteins
2.2.2.1. NAMI-A
2.2.2.2. KP1019
3. The Biological Profiles of NAMI-A and KP1019: Main Aspects
3.1. In Vitro Cellular Studies
3.2. Animal Tests and Biodistribution
3.3. Clinical Investigations
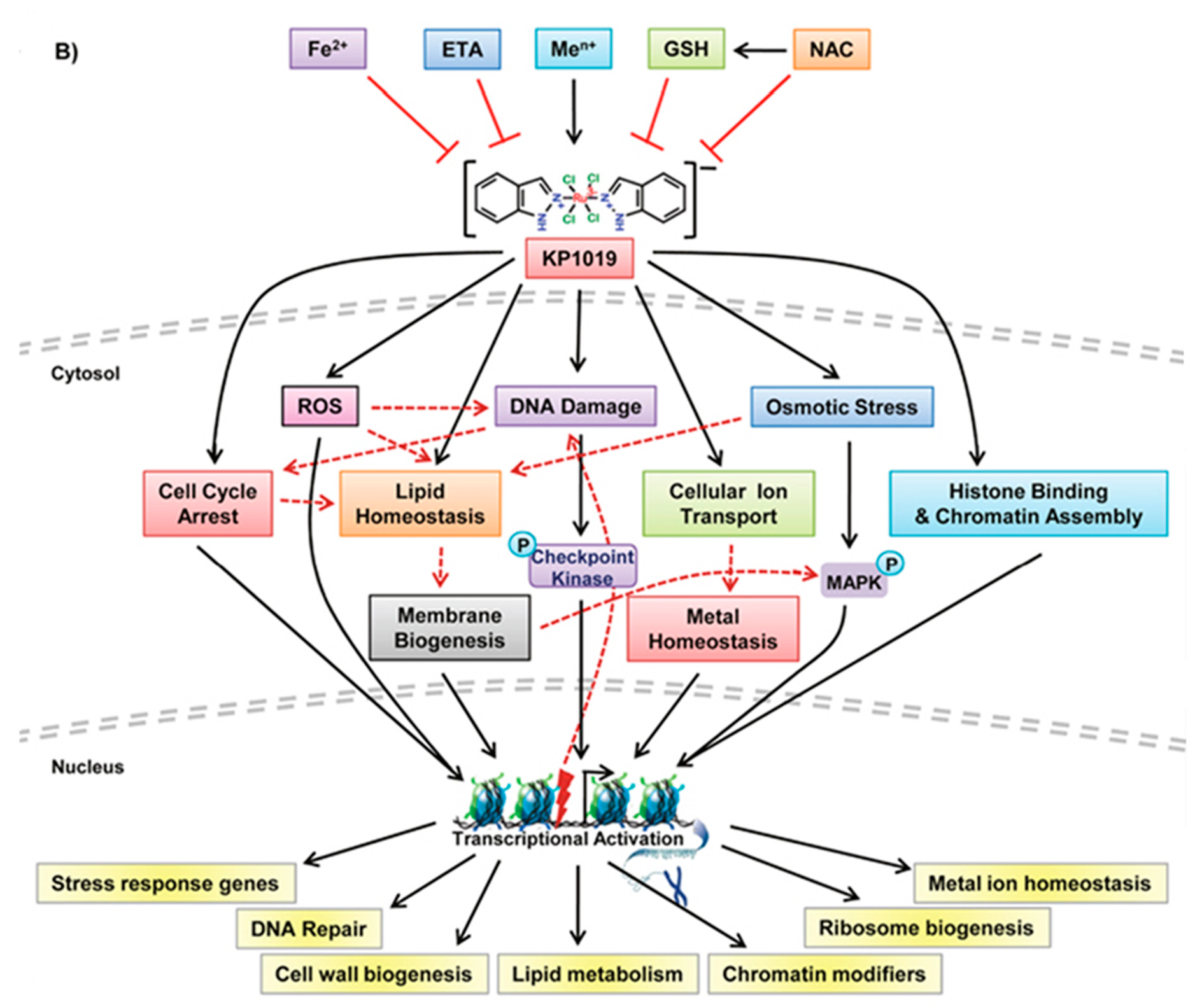
4. Mechanism(s) of Action: Myths and Facts
- Ruthenium (III) compounds are activated by reduction (activation-by-reduction mechanism)
- Ruthenium mimics iron metabolism and, as a consequence….
- …Transferrin is a selective carrier for ruthenium-based drugs, and….
- …Ruthenium compounds are not toxic
- DNA is the main target for ruthenium drugs.
- It is true that NAMI-A and KP1019/1339 are formally very similar. However, there are some apparently small but important chemical differences that heavily affect their respective reactivities and thus their biological profiles.
- Both Ru compounds are not very stable in physiological media, with NAMI-A being significantly less stable than KP1019. Upon considering their moderate stability under physiological conditions, both Ru drugs may be straightforwardly classified as prodrugs.
- Both compounds are sufficiently water-soluble and may be used as such.
- Both compounds enter the bloodstream and associate strongly to plasma proteins, in particular to serum albumin. In spite of that, substantial amounts of ruthenium distribute to the whole body and reach several body compartments in an effective way.
- Both compounds manifest a relatively low degree of systemic toxicity and may be used quite safely up to relevant concentrations.
- The pharmacological profile of the two is highly distinct: KP1019 still produces an appreciable cytotoxic effect, while NAMI-A mainly causes antimetastatic effects.
- The different behavior is probably mediated by their different interactions with cells. Indeed, KP1019 is able to enter cells in appreciable amounts, whereas NAMI-A mostly localizes extracellularly or on the cell membrane, this being in our opinion a very crucial mechanistic distinction.
4.1. NAMI-A
4.2. KP1019
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keppler, B.K.; Lipponer, K.-G.; Stenzel, B.; Kratz, F. New tumor-inhibiting ruthenium complexes In Metal Complexes in Cancer Chemotherapy; Keppler, B.K., Ed.; VCH: Weinheim, Germany, 1993; pp. 187–220. [Google Scholar]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A New Redox-Active Anticancer Agent – Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodiversity 2008, 5, 2140–2155. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef] [Green Version]
- Mestroni, G.; Alessio, E.; Sava, G.; Pacor, S.; Coluccia, M. The development of tumor-inhibiting ruthenium dimethylsulfoxide complexes. In Metal Complexes in Cancer Chemotherapy; Keppler, B.K., Ed.; VCH: Weinheim, Germany, 1993; pp. 157–185. [Google Scholar]
- Mestroni, G.; Alessio, E.; Sava, G.; Pacor, S.; Coluccia, M.; Boccarelli, A. Water soluble ruthenium(III)-dimethyl sulfoxide complexes: Chemical behaviour and pharmacological properties. Met.-Based Drugs 1994, 1, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E.; Mestroni, G.; Bergamo, A.; Sava, G. Ruthenium Anticancer Drugs. In Metal Ions in Biological Systems; Sigel, A., Sigel, H., Eds.; M. Dekker: New York, NY, USA, 2004; Volume 42, pp. 323–351. [Google Scholar]
- Alessio, E.; Mestroni, G.; Bergamo, A.; Sava, G. Ruthenium antimetastatic agents. Curr. Topics Med. Chem. 2004, 4, 1525–1535. [Google Scholar] [CrossRef]
- Bratsos, I.; Jedner, S.; Gianferrara, T.; Alessio, E. Ruthenium anticancer compounds: Challenges and expectations. Chimia 2007, 61, 692–697. [Google Scholar] [CrossRef]
- Bergamo, A.; Sava, G. Linking the future of anticancer metal-complexes to the therapy of tumour metastases. Chem. Soc. Rev. 2015, 44, 8818–8835. [Google Scholar] [CrossRef]
- Alessio, E. 30 years of the drug candidate NAMI-A and the myths in the field of ruthenium anticancer compounds: A personal perspective. Eur. J. Inorg. Chem. 2017, 1549–1560. [Google Scholar] [CrossRef]
- Sava, G.; Bergamo, A. Ruthenium-based compounds and tumor growth control. Int. J. Oncol. 2000, 17, 353–365. [Google Scholar]
- Kostova, I. Ruthenium Complexes as Anticancer Agents. Curr. Med. Chem. 2006, 13, 1085–1107. [Google Scholar]
- Jakupec, M.A.; Galanski, M.; Arion, V.B.; Hartinger, C.G.; Keppler, B.K. Antitumour metal compounds: More than theme and variations. Dalton Trans. 2008, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Emadi, A. Ruthenium-based chemotherapeutics: Are they ready for prime time? Cancer Chemother. Pharmacol. 2010, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bratsos, I.; Gianferrara, T.; Alessio, E.; Hartinger, C.G.; Jakupec, M.A.; Keppler, B.K. Ruthenium and Other Non-platinum Anticancer Compounds. In Bioinorganic Medicinal Chemistry; Alessio, E., Ed.; Wiley-VCH: Weinheim, Germany, 2011; pp. 151–174. [Google Scholar]
- Sava, G.; Bergamo, A.; Dyson, P.J. Metal-based antitumour drugs in the post-genomic era: What comes next? Dalton Trans. 2011, 40, 9069–9075. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Gaiddon, C.; Schellens, J.H.M.; Beijnen, J.H.; Sava, G. Approaching tumour therapy beyond platinum drugs. Status of the art and perspectives of ruthenium drug candidates. J. Inorg. Biochem. 2012, 106, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Medici, S.; Peana, M.; Nurchi, V.M.; Lachowicz, J.I.; Crisponi, G.; Zoroddu, M.A. Noble metals in medicine: Latest advances. Coord. Chem. Rev. 2015, 284, 329–350. [Google Scholar] [CrossRef]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure–activity relationships for ruthenium and osmium anticancer agents – towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]
- Coverdale, J.P.C.; Laroiya-McCarron, T.; Isolda Romero-Canelón, I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef]
- Clarke, M.J. Ruthenium metallopharmaceuticals. Coord. Chem. Rev. 2002, 232, 69–93. [Google Scholar]
- Keppler, B.K.; Rupp, W. Antitumor activity of imidazolium-bisimidazole-tetrachlororuthenate (III). J. Cancer Res. Clin. Oncol. 1986, 111, 166–168. [Google Scholar] [CrossRef]
- Berger, M.R.; Garzon, F.T.; Keppler, B.K.; Schmähl, D. Efficacy of new ruthenium complexes against chemically induced autochthonous colorectal carcinoma in rats. Anticancer Res. 1989, 9, 761–765. [Google Scholar] [PubMed]
- Peti, W.; Pieper, T.; Sommer, M.; Keppler, B.K.; Giester, G. Synthesis of Tumor-Inhibiting Complex Salts Containing the Anion trans-Tetrachlorobis(indazole)ruthenate(III) and Crystal Structure of the Tetraphenylphosphonium Salt. Eur. J. Inorg. Chem. 1999, 1551–1555. [Google Scholar]
- Alessio, E.; Balducci, G.; Calligaris, M.; Costa, G.; Attia, W.M.; Mestroni, G. Synthesis, molecular structure and chemical behavior of hydrogen trans-bis(dimethylsulfoxide)tetrachlororutenate(III) and mer-trichlorotris(dimethylsulfoxide)ruthenium(III): The first fully characterized chloro-dimethylsulfoxide-ruthenium(III) complexes. Inorg. Chem. 1991, 30, 609–618. [Google Scholar] [CrossRef]
- Alessio, E.; Balducci, G.; Lutman, A.; Mestroni, G.; Calligaris, M.; Attia, W.M. Synthesis and characterization of two new classes of ruthenium(III)-sulfoxide complexes with nitrogen donor ligands (L): Na[trans-RuCl4(R2SO)(L)] and mer,cis-RuCl3(R2SO)(R2SO)(L). The crystal structure of Na[trans-RuCl4(DMSO)(NH3)]·2DMSO, Na[trans-RuCl4(DMSO)(Im)]·H2O·Me2CO (Im = imidazole) and mer,cis- RuCl3(DMSO)(DMSO)(NH3). Inorg. Chim. Acta 1993, 203, 205–217. [Google Scholar]
- Sava, G.; Pacor, S.; Mestroni, G.; Alessio, E. Na[trans-RuCl4(DMSO)Im], a metal complex of ruthenium with antimetastatic properties. Clin. Exp. Metastasis 1992, 10, 273–280. [Google Scholar]
- Sava, G.; Pacor, S.; Mestroni, G.; Alessio, E. Effects of the Ru(III) complexes mer-RuCl3(DMSO)2Im and Na[trans-RuCl4(DMSO)Im] on solid mouse tumors. Anti-Cancer Drugs 1992, 3, 25–31. [Google Scholar] [CrossRef]
- Galeano, A.; Berger, M.R.; Keppler, B.K. Antitumor activity of some ruthenium derivatives in human colon cancer cell lines in vitro. Arzneimittel-Forschung/Drug Res. 1992, 42, 821–824. [Google Scholar]
- Bouma, M.; Nuijen, B.; Jansen, M.T.; Sava, G.; Flaibani, A.; Bult, A.; Beijnen, J.H. A kinetic study of the chemical stability of the antimetastatic ruthenium complex NAMI-A. Int. J. Pharm. 2002, 248, 239–246. [Google Scholar]
- Bacac, M.; Hotze, A.C.G.; van der Schilden, K.; Haasnoot, J.G.; Pacor, S.; Alessio, E.; Sava, G.; Reedijk, J. The hydrolysis of the anti-cancer ruthenium complex NAMI-A affects its DNA binding and antimetastatic activity: An NMR evaluation. J. Inorg. Biochem. 2004, 98, 402–412. [Google Scholar] [CrossRef]
- Bergamo, A.; Gava, B.; Alessio, E.; Mestroni, G.; Serli, B.; Cocchietto, M.; Zorzet, S.; Sava, G. Ruthenium-based NAMI-A type complexes with in vivo selective metastasis reduction and in vitro invasion inhibition unrelated to cell cytotoxicity. Int. J. Oncol. 2002, 21, 1331–1338. [Google Scholar] [CrossRef]
- Sava, G.; Bergamo, A.; Zorzet, S.; Gava, B.; Casarsa, C.; Cocchietto, M.; Furlani, A.; Scarcia, V.; Serli, B.; Iengo, E.; et al. Influence of chemical stability on the activity of the antimetastasis ruthenium compound NAMI-A. Eur. J. Cancer 2002, 38, 427–435. [Google Scholar] [CrossRef]
- Reisner, E.; Arion, V.B.; Guedes da Silva, M.F.C.; Lichtenecker, R.; Eichinger, A.; Keppler, B.K.; Kukushkin, V.Y.; Pombeiro, A.J.L. Tuning of Redox Potentials for the Design of Ruthenium Anticancer Drugs—An Electrochemical Study of [trans-RuCl4L(DMSO)]– and [trans-RuCl4L2]– Complexes, where L = Imidazole, 1,2,4-Triazole, Indazole. Inorg. Chem. 2004, 43, 7083–7093. [Google Scholar] [CrossRef] [PubMed]
- Brindell, M.; Piotrowska, D.; Shoukry, A.A.; Stochel, G.; van Eldik, R. Kinetics and mechanism of the reduction of (ImH)[trans-RuCl4(dmso)(Im)] by ascorbic acid in acidic aqueous solution. J. Biol. Inorg. Chem. 2007, 12, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Küng, A.; Pieper, T.; Wissiack, R.; Rosenberg, E.; Keppler, B.K. Hydrolysis of the tumor-inhibiting ruthenium(III) complexes HIm[trans-RuCl4(Im)2] and HInd[trans-RuCl4(Ind)2] investigated by means of HPCE and HPLC-MS. J. Biol. Inorg. Chem. 2001, 6, 292–299. [Google Scholar]
- Cebrian-Losantos, B.; Reisner, E.; Kowol, C.R.; Roller, A.; Shova, S.; Arion, V.B.; Keppler, B.K. Synthesis and Reactivity of the Aquation Product of the Antitumor Complex trans-[RuIIICl4(indazole)2]–. Inorg. Chem. 2008, 47, 6513–6523. [Google Scholar] [CrossRef]
- Cetinbas, N.; Webb, M.I.; Dubland, J.A.; Walsby, C.J. Serum-protein interactions with anticancer Ru(III) complexes KP1019 and KP418 characterized by EPR. J. Biol. Inorg. Chem. 2010, 15, 131–145. [Google Scholar] [CrossRef]
- Schluga, P.; Hartinger, C.G.; Egger, A.; Reisner, E.; Galanski, M.; Jakupec, M.A.; Keppler, B.K. Redox behavior of tumor-inhibiting ruthenium(III) complexes and effects of physiological reductants on their binding to GMP. Dalton Trans. 2006, 1796–1802. [Google Scholar] [CrossRef]
- Malina, J.; Novakova, O.; Keppler, B.K.; Alessio, E.; Brabec, V. Biophysical analysis of natural, double-helical DNA modified by anticancer heterocyclic complexes of ruthenium(III) in cell-free media. J. Biol. Inorg. Chem. 2001, 6, 435–445. [Google Scholar] [CrossRef]
- Brabec, V.; Kasparkova, J. Ruthenium coordination compounds of biological and biomedical significance. DNA binding agents. Coord. Chem. Rev. 2018, 376, 75–94. [Google Scholar] [CrossRef]
- Groessl, M.; Tsybin, Y.O.; Hartinger, C.G.; Keppler, B.K.; Dyson, P.J. Ruthenium versus platinum: Interactions of anticancer metallodrugs with duplex oligonucleotides characterised by electrospray ionisation mass spectrometry. J. Biol. Inorg. Chem. 2010, 15, 677–688. [Google Scholar] [CrossRef]
- Musumeci, D.; Rozza, L.; Merlino, A.; Paduano, L.; Marzo, T.; Massai, L.; Messori, L.; Montesarchio, D. Interaction of anticancer Ru(III) complexes with single stranded and duplex DNA model systems. Dalton Trans. 2015, 44, 13914–13925. [Google Scholar] [CrossRef]
- Hostetter, A.A.; Miranda, M.L.; DeRose, V.J.; Holman, K.L. McFarlane Holman, Ru binding to RNA following treatment with the antimetastatic prodrug NAMI-A in Saccharomyces cerevisiae and in vitro. J. Biol. Inorg. Chem. 2011, 16, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, B.G.; Johnson, E.; Cazares, E.; McFarlane Holman, K.L.; Kirk, S.R. Ruthenium anticancer agent KP1019 binds more tightly than NAMI-A to tRNAPhe. J. Inorg. Bichem. 2018, 182, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Luck, A.N.; Mason, A.B. Structure and Dynamics of Drug Carriers and Their Interaction with Cellular Receptors: Focus on Serum Transferrin. Adv. Drug Deliv. Rev. 2013, 65, 1012–1019. [Google Scholar] [CrossRef]
- Kratz, F.; Beyer, U. Serum proteins as drug carriers of anticancer agents: A review. Drug Deliv. 1998, 5, 281–299. [Google Scholar] [CrossRef]
- Daniels, T.R.; Bernabeu, E.; Rodríguez, J.A.; Patel, S.; Kozman, M.; Chiappetta, D.A.; Holler, E.; Ljubimova, J.Y.; Helguera, G.; Penichet, M.L. Transferrin receptors and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta. 2012, 1820, 291–317. [Google Scholar] [CrossRef]
- Levina, A.; Aitken, J.B.; Gwee, Y.Y.; Lim, Z.J.; Liu, M.; Singharay, A.M.; Wong, P.F.; Lay, P.A. Biotransformations of Anticancer Ruthenium(III) Complexes: An X-Ray Absorption Spectroscopic Study. Chem. Eur. J. 2013, 19, 3609–3619. [Google Scholar] [CrossRef]
- Webb, M.I.; Walsby, C.J. Control of ligand-exchange processes and the oxidation state of the antimetastatic Ru(III) complex NAMI-A by interactions with human serum albumin. Dalton Trans. 2011, 40, 1322–1331. [Google Scholar] [CrossRef]
- Webb, M.I.; Walsby, C.J. EPR as a probe of the intracellular speciation of ruthenium(III) anticancer compounds. Metallomics 2013, 5, 1624–1633. [Google Scholar] [CrossRef]
- Webb, M.I.; Walsby, C.J. Albumin binding and ligand-exchange processes of the Ru(III) anticancer agent NAMI-A and its bis-DMSO analogue determined by ENDOR spectroscopy. Dalton Trans. 2015, 44, 17482–17493. [Google Scholar] [CrossRef]
- Gransbury, G.K.; Kappen, P.; Glover, C.J.; Hughes, J.N.; Levina, A.; Lay, P.A.; Musgrave, I.F.; Harris, H.H. Comparison of KP1019 and NAMI-A in tumour-mimetic environments. Metallomics 2016, 8, 762–773. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Lim, Z.J.; Gwee, Y.Y.; Levina, A.; Lay, P.A. Characterization of a Ruthenium(III)/NAMI-A Adduct with Bovine Serum Albumin that Exhibits a High Anti-Metastatic Activity. Angew. Chem. Int. Ed. Engl. 2010, 49, 1661–1664. [Google Scholar] [CrossRef] [PubMed]
- Novohradský, V.; Bergamo, A.; Cocchietto, M.; Zajac, J.; Brabec, V.; Mestroni, G.; Sava, G. Influence of the binding of reduced NAMI-A to human serum albumin on the pharmacokinetics and biological activity. Dalton Trans. 2015, 44, 1905–1913. [Google Scholar]
- Merlino, A. Interactions between proteins and Ru compounds of medicinal interest: A structural perspective. Coord. Chem. Rev. 2016, 326, 111–134. [Google Scholar] [CrossRef]
- Messori, L.; Merlino, A. Ruthenium metalation of proteins: The X-ray structure of the complex formed between NAMI-A and hen egg white lysozyme. Dalton Trans. 2014, 43, 6128–6131. [Google Scholar]
- Casini, A.; Temperini, C.; Gabbiani, C.; Supuran, C.T.; Messori, L. The X-ray structure of the adduct between NAMI-A and carbonic anhydrase provides insights into the reactivity of this metallodrug with proteins. ChemMedChem 2010, 5, 1989–1994. [Google Scholar] [CrossRef] [PubMed]
- Ciambellotti, S.; Pratesi, A.; Severi, M.; Ferraro, G.; Alessio, E.; Merlino, A.; Messori, L. The NAMI A – Human Ferritin System: A Biophysical Characterization. Dalton Trans. 2018, 47, 11429–11437. [Google Scholar] [CrossRef] [PubMed]
- Vergara, A.; D’Errico, G.; Montesarchio, D.; Mangiapia, G.; Paduano, L.; Merlino, A. Interaction of Anticancer Ruthenium Compounds with Proteins: High-Resolution X-ray Structures and Raman Microscopy Studies of the Adduct between Hen Egg White Lysozyme and AziRu. Inorg. Chem. 2013, 52, 4157–4159. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, C.; Musumeci, D.; Irace, C.; Paduano, L.; Montesarchio, D. RuIII Complexes for Anticancer Therapy: The Importance of Being Nucleolipidic. Eur. J. Org. Chem. 2017, 1100–1119. [Google Scholar] [CrossRef]
- Smith, C.A.; Sutherland-Smith, A.J.; Keppler, B.K.; Kratz, F.; Baker, E.N. Binding of ruthenium(III) anti-tumor drugs to human lactoferrin probed by high resolution X-ray crystallographic structure analyses. J. Biol. Inorg. Chem. 1996, 1, 424–431. [Google Scholar] [CrossRef]
- Bijelic, A.; Theiner, S.; Keppler, B.K.; Rompel, A. X-ray Structure Analysis of Indazolium trans-[Tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019) Bound to Human Serum Albumin Reveals Two Ruthenium Binding Sites and Provides Insights into the Drug Binding Mechanism. J. Med. Chem. 2016, 59, 5894–5903. [Google Scholar] [CrossRef]
- Sava, G.; Pacor, S.; Bergamo, A.; Cocchietto, M.; Mestroni, G.; Alessio, E. Effects of ruthenium complexes on experimental tumors: Irrelevance of cytotoxicity for metastasis inhibition. Chem.-Biol. Interactions 1995, 95, 109–126. [Google Scholar] [CrossRef]
- Bergamo, A.; Gagliardi, R.; Scarcia, V.; Furlani, A.; Alessio, E.; Mestroni, G.; Sava, G. In vitro cell cycle arrest, in vivo action on solid metastasizing tumors and host toxicity of the antimetastatic drug NAMI-A and of cisplatin. J. Pharmacol. Exp. Ther. 1999, 289, 559–564. [Google Scholar]
- Pluim, D.; van Waardenburg, R.C.A.M.; Beijnen, J.H.; Schellens, J.H.M. Cytotoxicity of the organic ruthenium anticancer drug Nami-A is correlated with DNA binding in four different human tumor cell lines. Cancer Chemother. Pharmacol. 2004, 54, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.; Capozzi, I.; Clerici, K.; Gagliardi, G.; Alessio, E.; Mestroni, G. Pharmacological control of lung metastases of solid tumors by a novel ruthenium complex. Clin. Exp. Metastasis. 1998, 16, 371–379. [Google Scholar] [CrossRef]
- Pillozzi, S.; Gasparoli, L.; Stefanini, M.; Ristori, M.; D’Amico, M.; Alessio, E.; Scaletti, F.; Becchetti, A.; Arcangeli, A.; Messori, L. NAMI-A is highly cytotoxic toward leukaemia cell lines: Evidence of inhibition of KCa 3.1 channels. Dalton Trans. 2014, 43, 12150–12155. [Google Scholar] [CrossRef]
- Heffeter, P.; Pongratz, M.; Steiner, E.; Chiba, P.; Jakupec, M.A.; Elbling, L.; Marian, B.; Körner, W.; Sevelda, F.; Micksche, M.; et al. Intrinsic and Acquired Forms of Resistance against the Anticancer Ruthenium Compound KP1019 [Indazolium trans-[tetrachlorobis(1H-indazole)ruthenate (III)] (FFC14A). J. Pharmacol. Exp. Ther. 2005, 312, 281–289. [Google Scholar] [CrossRef]
- Heffeter, P.; Böck, K.; Atil, B.; Hoda, M.A.R.; Körner, W.; Bartel, C.; Jungwirth, U.; Keppler, B.K.; Micksche, M.; Berger, W.; et al. Intracellular protein binding patterns of the anticancer ruthenium drugs KP1019 and KP1339. J. Biol. Inorg. Chem. 2010, 15, 737–748. [Google Scholar] [CrossRef] [Green Version]
- Kapitza, S.; Pongratz, M.; Jakupec, M.A.; Heffeter, P.; Berger, W.; Lackinger, L.; Keppler, B.K.; Marian, B. Heterocyclic complexes of ruthenium(III) induce apoptosis in colorectal carcinoma cells. J. Cancer Res. Clin. Oncol. 2005, 131, 101–110. [Google Scholar] [CrossRef]
- Schreiber-Brynzak, E.; Klapproth, E.; Unger, C.; Lichtscheidl-Schultz, I.; Goeschl, S.; Schweighofer, S.; Trondl, R.; Dolznig, H.; Jakupec, M.A.; Keppler, B.K. Three-dimensional and co-culture models for preclinical evaluation of metal-based anticancer drugs. Investig. New Drugs 2015, 33, 835–847. [Google Scholar] [CrossRef]
- Bergamo, A.; Masi, A.; Jakupec, M.A.; Keppler, B.K.; Sava, G. Inhibitory Effects of the Ruthenium Complex KP1019 in Models of Mammary Cancer Cell Migration and Invasion. Met.-Based Drugs 2009, 681270. [Google Scholar] [CrossRef]
- Coluccia, M.; Sava, G.; Salerno, G.; Bergamo, A.; Pacor, S.; Mestroni, G.; Alessio, E. Efficacy of 5-FU combined to Na[trans-RuCl4(DMSO)(Im)], a novel selective antimetastatic agent, on the survival time of mice with P388 leukemia, P388/DDP subline and MCa mammary carcinoma. Met. -Based Drugs 1995, 2, 195–199. [Google Scholar] [CrossRef]
- Sava, G.; Zorzet, S.; Turrin, C.; Vita, F.; Soranzo, M.R.; Zabucchi, G.; Cocchietto, M.; Bergamo, A.; DiGiovine, S.; Pezzoni, G.; et al. Dual Action of NAMI-A in Inhibition of Solid Tumor Metastasis: Selective Targeting of Metastatic Cells and Binding to Collagen. Clin. Cancer Res. 2003, 9, 1898–1905. [Google Scholar]
- Gava, B.; Zorzet, S.; Spessotto, P.; Cocchietto, M.; Sava, G. Inhibition of B16 Melanoma Metastases with the Ruthenium Complex Imidazolium trans-Imidazoledimethylsulfoxide-tetrachlororuthenate and Down-Regulation of Tumor Cell Invasion. J. Pharmacol. Exp. Ther. 2006, 317, 284–291. [Google Scholar] [CrossRef]
- Groessl, M.; Reisner, E.; Hartinger, C.G.; Eichinger, R.; Semenova, O.; Timerbaev, A.R.; Jakupec, M.A.; Arion, V.B.; Keppler, B.K. Structure-Activity Relationships for NAMI-A-type Complexes (HL)[trans-RuCl4L(S-dmso)ruthenate(III)] (L = Imidazole, Indazole, 1,2,4-Triazole, 4-Amino-1,2,4-triazole, and 1-Methyl-1,2,4-triazole): Aquation, Redox Properties, Protein Binding, and Antiproliferative Activity. Activity J. Med. Chem. 2007, 50, 2185–2193. [Google Scholar]
- Bergamo, A.; Sava, G. Ruthenium complexes can target determinants of tumour malignancy. Dalton Trans. 2007, 1267–1272. [Google Scholar] [CrossRef]
- Zorzet, S.; Bergamo, A.; Cocchietto, M.; Sorc, A.; Gava, B.; Alessio, E.; Iengo, E.; Sava, G. Lack of in vitro cytotoxicity, associated to increased G2-M cell fraction and inhibition of matrigel invasion, may predict in vivo-selective antimetastasis activity of ruthenium complexes. J. Pharmacol. Exp. Ther. 2000, 295, 927–933. [Google Scholar]
- Pacor, S.; Zorzet, S.; Cocchietto, M.; Bacac, M.; Vadori, M.; Turrin, C.; Gava, B.; Castellarin, A.; Sava, G. Intratumoral NAMI-A Treatment Triggers Metastasis Reduction, Which Correlates to CD44 Regulation and Tumor Infiltrating Lymphocyte Recruitment. J. Pharmacol. Exp. Ther. 2004, 310, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Seelig, M.H.; Berger, M.R.; Keppler, B.K. Antineoplastic activity of three ruthenium derivatives against chemically induced colorectal carcinoma in rats. J. Cancer Res. Clin. Oncol. 1992, 118, 195–200. [Google Scholar] [CrossRef]
- Depenbrock, H.; Schmelcher, S.; Peter, R.; Keppler, B.K.; Weirich, G.; Block, T.; Rastetter, J.; Hanauske, A.R. Preclinical Activity of Trans-indazolium [Tetrachlorobisindazoleruthenate(III)] (NSC 666158; IndCR.; KP 1019) Against Tumour Colony-forming Units and Haematopoietic Progenitor Cells. Cells Eur. J. Cancer 1997, 33, 2404–2410. [Google Scholar] [CrossRef]
- Bytzek, A.K.; Koellensperger, G.; Keppler, B.K.; Hartinger, C.G. Biodistribution of the novel anticancer drug sodium trans-[tetrachloridobis(1H-indazole)ruthenate(III)] KP-1339/IT139 in nude BALB/c mice and implications on its mode of action. J. Inorg. Biochem. 2016, 160, 250–255. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluil, D.; Beijnen, J.H.; Schellens, J.H.M. A Phase I and Pharmacological Study with Imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a Novel Ruthenium Anticancer Agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [PubMed]
- Cocchietto, M.; Sava, G. Blood concentration and toxicity of the antimetastasis agent NAMI-A following repeated intravenous treatment in mice. Pharmacol. Toxicol. 2000, 87, 193–197. [Google Scholar] [CrossRef]
- Jassem, J.; Krzakowski, M.; Roszkowski, K.; Ramlau, R.; Słomiński, J.M.; Szczęsna, A.; Krawczyk, K.; Moźejko-Pastewka, B.; Lis, J.; Miracki, K. A phase II study of gemcitabine plus cisplatin in patients with advanced non-small cell lung cancer: Clinical outcomes and quality of life. Lung Cancer 2002, 35, 73–79. [Google Scholar] [CrossRef]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Investig. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; Van, G.M.; van Oosterom, A.T.; Christian, M.C.; et al. New guidelines to evaluate the response to treatment in solid tumors. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside – preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Henke, M.M.; Richly, H.; Drescher, A.; Grubert, M.; Alex, D.; Thyssen, D.; Jaehde, U.; Scheulen, M.E.; Hilger, R.A. Pharmacokinetic study of sodium trans[tetrachlorobis(1H-indazole)-ruthenate (III)]/-indazole hydrochloride (1:1.1) (FFC14A) in patients with solid tumors. Int. J. Clin. Pharmacol. Ther. 2009, 47, 58–60. [Google Scholar] [CrossRef]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef]
- Fuereder, T.; Berger, W. Metal drugs become targeted. ESMO Open 2017, 2, e000239. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Dyson, P.J.; Sava, G. The mechanism of tumour cell death by metal-based anticancer drugs is not only a matter of DNA interactions. Coord. Chem. Rev. 2018, 360, 17–33. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs: A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef]
- Englinger, B.; Pirker, C.; Heffeter, P.; Terenzi, A.; Kowol, C.R.; Keppler, B.K.; Berger, W. Metal Drugs and the Anticancer Immune Response. Chem. Rev. 2019, 119, 1519–1624. [Google Scholar] [CrossRef]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T.A. Subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.-S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef]
- Pal, M.; Nandi, U.; Mukherjee, D. Detailed account on activation mechanisms of ruthenium coordination complexes and their role as antineoplastic agents. Eur. J. Med. Chem. 2018, 150, 419–445. [Google Scholar] [CrossRef]
- Blazevic, A.; Hummer, A.A.; Heffeter, P.; Berger, W.; Filipits, M.; Cibin, G.; Keppler, B.K.; Rompel, A. Electronic State of Sodium trans-[Tetrachloridobis(1H-indazole)ruthenate(III)] (NKP-1339) in Tumor, Liver and Kidney Tissue of a SW480-bearing Mouse. Sci. Rep. 2017, 7, 40966. [Google Scholar] [CrossRef]
- Sulyok, M.; Hann, S.; Hartinger, C.G.; Keppler, B.K.; Stingeder, G.; Koellensperger, G. Two dimensional separation schemes for investigation of the interaction of an anticancer ruthenium(III) compound with plasma proteins. J. Anal. At. Spectrom. 2005, 20, 856–863. [Google Scholar] [CrossRef]
- Groessl, M.; Hartinger, C.G.; Polec-Pawlak, K.; Jarosz, M.; Keppler, B.K. Capillary electrophoresis hyphenated to inductively coupled plasma-mass spectrometry: A novel approach for the analysis of anticancer metallodrugs in human serum and plasma. Electrophoresis 2008, 29, 2224–2232. [Google Scholar] [CrossRef]
- Dömötör, O.; Hartinger, C.G.; Bytzek, A.K.; Kiss, T.; Keppler, B.K.; Enyedy, E. Characterization of the binding sites of the anticancer ruthenium(III) complexes KP1019 and KP1339 on human serum albumin via competition studies. J. Biol. Inorg. Chem. 2013, 18, 9–17. [Google Scholar]
- Śpiewak, K.; Brindell, M. Impact of low- and high-molecular-mass components of human serum on NAMI-A binding to transferrin. J. Biol. Inorg. Chem. 2015, 20, 695–703. [Google Scholar] [Green Version]
- Pelillo, C.; Mollica, H.; Eble, J.A.; Grosche, J.; Herzog, L.; Codan, B.; Sava, G.; Bergamo, A. Inhibition of adhesion, migration and of α5β1 integrin in the HCT-116 colorectal cancer cells treated with the ruthenium drug NAMI-A. J. Inorg. Biochem. 2016, 160, 225–235. [Google Scholar] [CrossRef]
- Sava, G.; Frausin, F.; Cocchietto, M.; Vita, F.; Podda, E.; Spessotto, P.; Furlani, A.; Scarcia, V.; Zabucchi, G. Actin-dependent tumour cell adhesion after short-term exposure to the antimetastasis ruthenium complex NAMI-A. Eur. J. Cancer 2004, 40, 1383–1396. [Google Scholar] [CrossRef]
- Casarsa, C.; Mischis, M.T.; Sava, G. TGFβ1 regulation and collagen-release-independent connective tissue re-modelling by the ruthenium complex NAMI-A in solid tumours. J. Inorg. Biochem. 2004, 98, 1648–1654. [Google Scholar] [CrossRef]
- Aitken, J.B.; Antony, S.; Weekley, C.M.; Lai, B.; Spiccia, L.; Harris, H.H. Distinct cellular fates for KP1019 and NAMI-A determined by X-ray fluorescence imaging of single cells. Metallomics 2012, 4, 1051–1056. [Google Scholar] [CrossRef]
- Vacca, A.; Bruno, M.; Boccarelli, A.; Coluccia, M.; Ribatti, D.; Bergamo, A.; Garbisa, S.; Sartor, L.; Sava, G. Inhibition of endothelial cell functions and of angiogenesis by the metastasis inhibitor NAMI-A. Br. J. Cancer 2002, 86, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Morbidelli, L.; Donnini, S.; Filippi, S.; Messori, L.; Piccioli, F.; Orioli, P.; Sava, G.; Ziche, M. Antiangiogenic properties of selected ruthenium(III) complexes that are nitric oxide scavengers. Br. J. Cancer 2003, 88, 1484–1491. [Google Scholar] [CrossRef] [Green Version]
- Flocke, L.S.; Trondl, R.; Jakupec, M.A.; Keppler, B.K. Molecular mode of action of NKP-1339 – a clinically investigated ruthenium-based drug—involves ER- and ROS-related effects in colon carcinoma cell lines. Investig. New Drugs 2016, 34, 261–268. [Google Scholar] [CrossRef]
- Golla, U.; Swagatika, S.; Chauhan, S.; Tomar, R.S. A systematic assessment of chemical, genetic, and epigenetic factors influencing the activity of anticancer drug KP1019 (FFC14A). Oncotarget 2017, 8, 98426–98454. [Google Scholar] [CrossRef] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alessio, E.; Messori, L. NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry. Molecules 2019, 24, 1995. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101995
Alessio E, Messori L. NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry. Molecules. 2019; 24(10):1995. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101995
Chicago/Turabian StyleAlessio, Enzo, and Luigi Messori. 2019. "NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry" Molecules 24, no. 10: 1995. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101995