The Development of a qPCR Assay to Measure Aspergillus flavus Biomass in Maize and the Use of a Biocontrol Strategy to Limit Aflatoxin Production


Abstract
:1. Introduction
2. Results
2.1. Colonisation of Plant Yissues by A. flavus
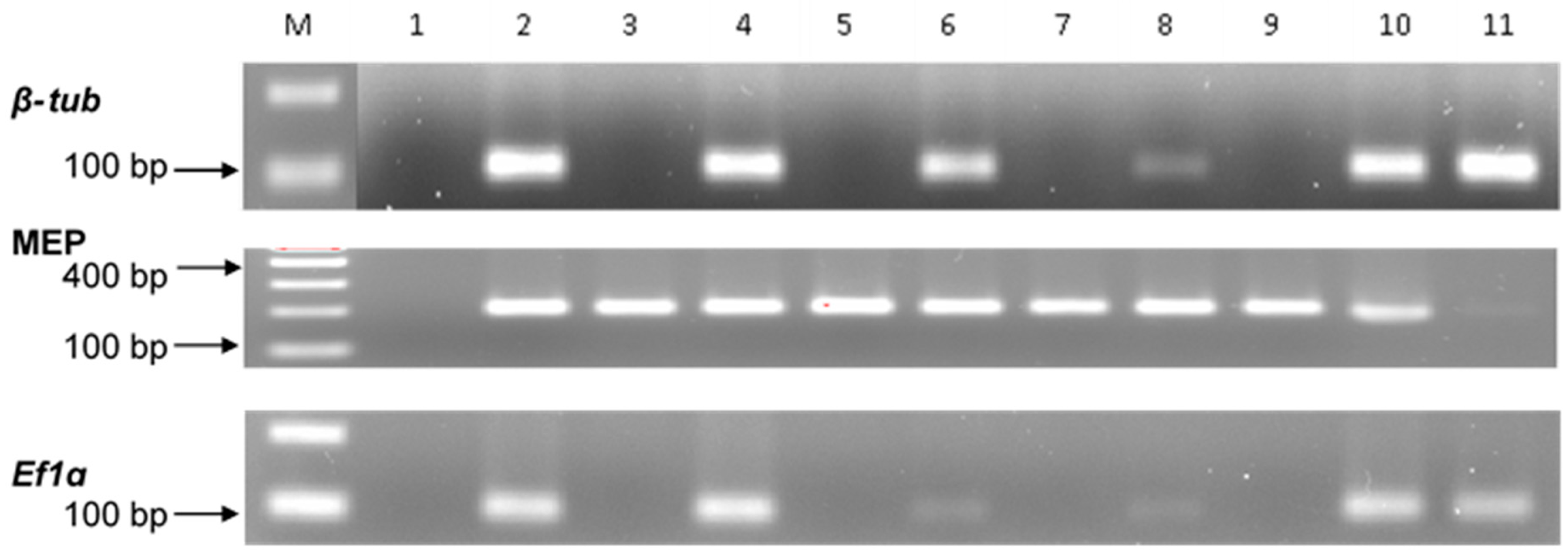
2.2. Gene Specificity and RT-qPCR Assays
2.3. In-Vitro Viocontrol Strategies in Aflatoxin Management and Aspergillus Flavus
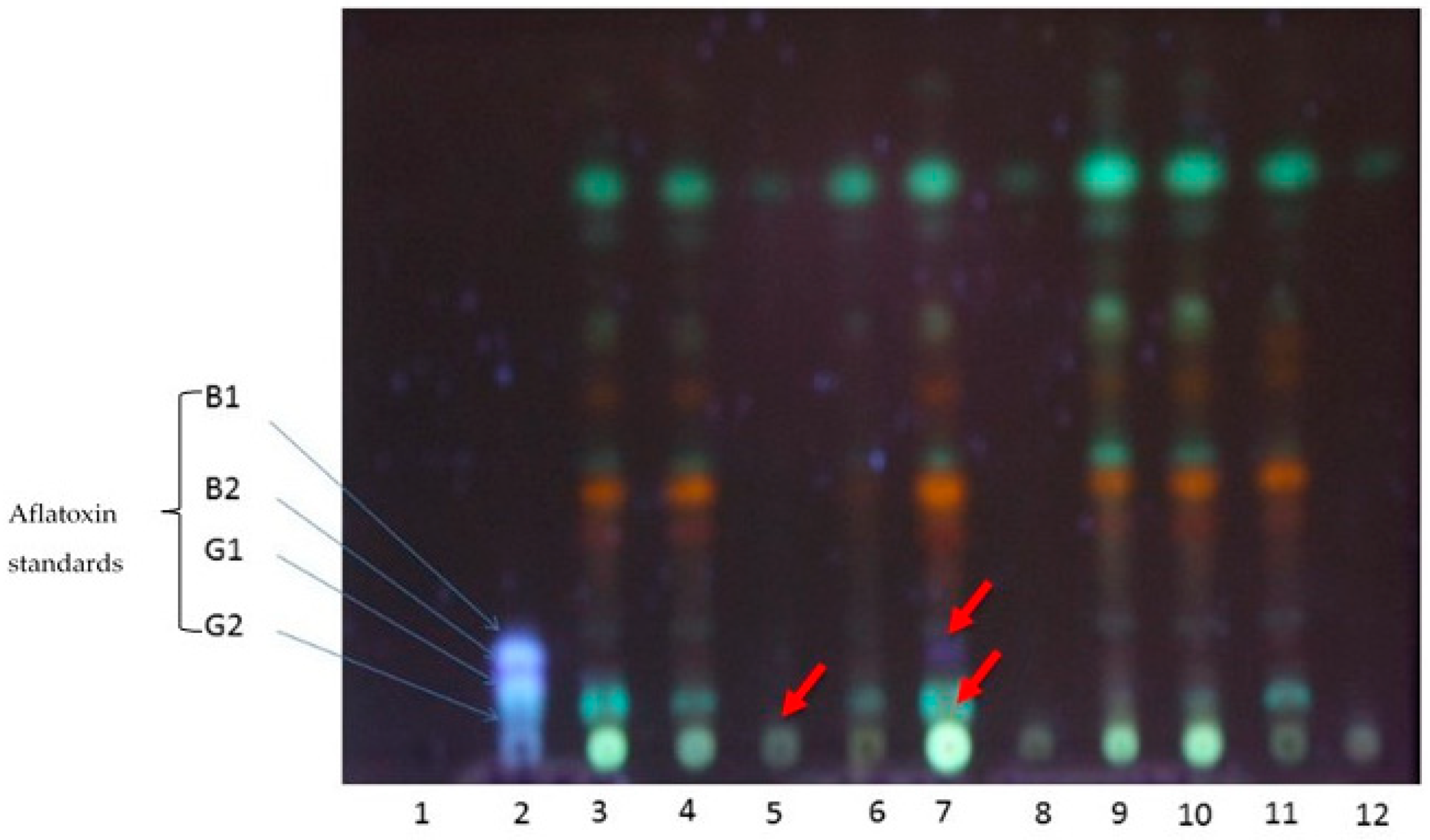
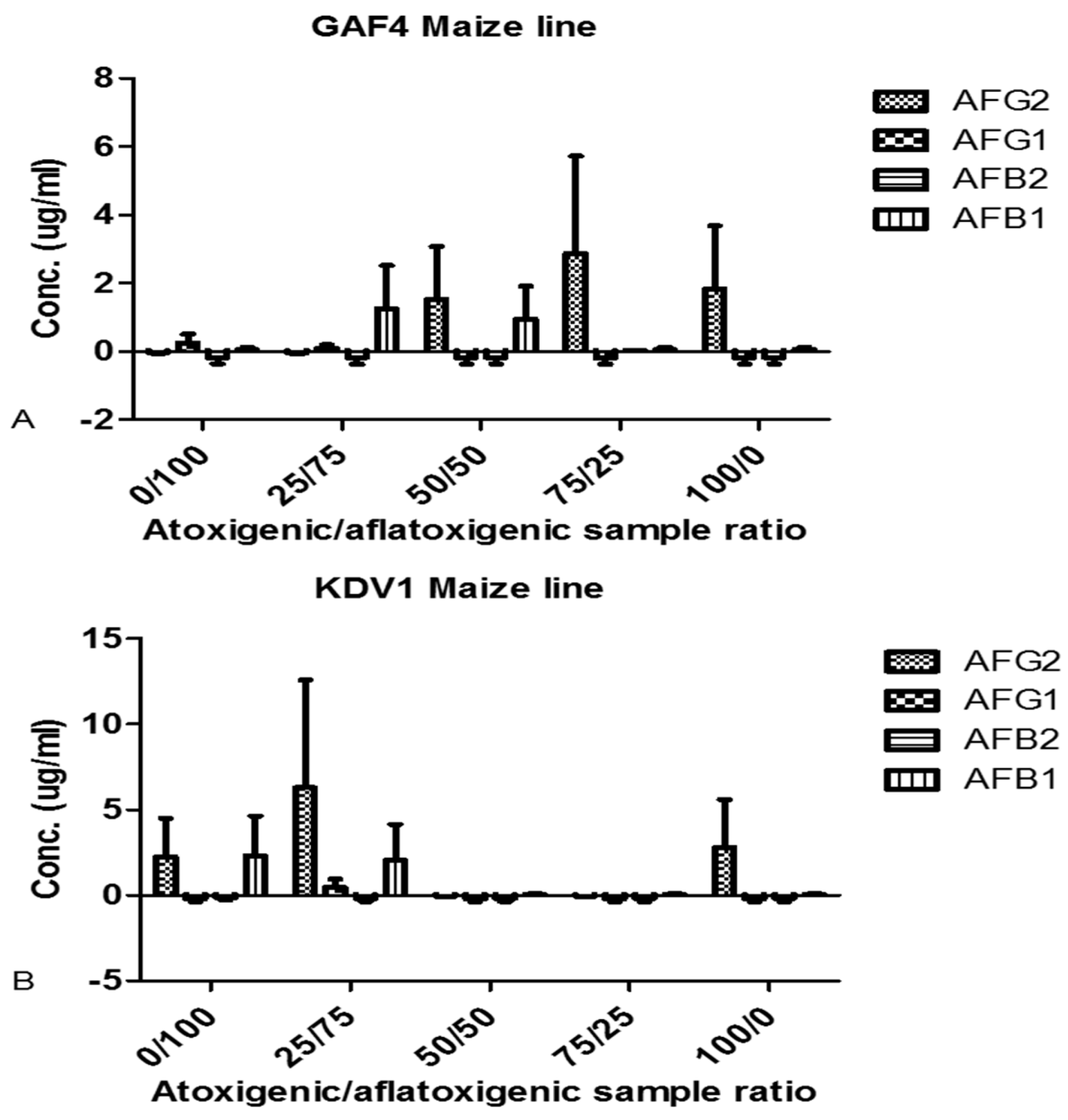
2.4. Aflatoxin Analyses after Biocontrol Strategy
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cultures of Fungi
5.2. Maize Cultivars
5.3. Reagents and Media Preparation
5.4. Seed Sterilisation and Aspergillus flavus Infection−
5.5. DNA Extraction from Aspergillus flavus and Maize Tissues
5.6. Primer Design
5.7. PCR Amplification
5.8. Aflatoxin Standards, Standard Curves and Fungal Quantification
5.9. Metabolite Extraction
5.10. Thin Layer Chromatography
- E = aflatoxins (µg/kg)
- S = µL of aflatoxin standard equal in fluorescence to sample spot
- C = aflatoxin standard concentration in µg/mL
- V = final dilution of the sample extract (µL)
- Z = sample matching the standards (µL)
- W = sample extract weight (mg)
5.11. High Performance Liquid Chromatography and Optimisation
5.12. In Vitro Co-Infection of Maize Lines and Biocontrol Strategy
5.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bandyopadhyay, R.; Kiewnick, S.; Atehnkeng, J.; Matthias, D.; Cottyc, P.J.; Kerstin, H. Biological control of aflatoxin contamination in maize in Africa. In Proceedings of the Conference on International Agricultural Research for Development, Stuttgart, Germany, 11–13 October 2005; pp. 1–8. [Google Scholar]
- Frisvad, J.C.; Larsen, T.O.; De Vries, R.; Meijer, M.; Houbraken, J.; Cabañes, F.J.; Ehrlich, K.; Samson, R.A. Secondary metabolite profiling, growth profiles and other tools for species recognition and important Aspergillus mycotoxins. Stud. Mycol. 2007, 59, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Leger, R.J.S.T.; Screen, S.E.; Shams-pirzadeh, B.; Rica, C. Lack of Host Specialization in Aspergillus flavus. Appl. Environ. Microbiol. 2000, 66, 320–324. [Google Scholar] [CrossRef]
- Pitt, J.I.; Taniwaki, M.H.; Cole, M.B. Mycotoxin production in major crops as influenced by growing, harvesting, storage and processing, with emphasis on the achievement of Food Safety Objectives. Food Control 2013, 32, 205–215. [Google Scholar] [CrossRef]
- Reddy, K.R.N.; Reddy, C.S.; Muralidharan, K. Potential of botanicals and biocontrol agents on growth and aflatoxin production by Aspergillus flavus infecting rice grains. Food Control 2009, 20, 173–178. [Google Scholar] [CrossRef]
- Ngindu, A.; Kenya, P.; Ocheng, D.; Omondi, T.; Ngare, W.; Gatei, D.; Johnson, B.; Ngira, J.; Nandwa, H.; Jansen, A.; et al. Outbreak of acute hepatitis caused by aflatoxin poisoning in Kenya. Lancet 1982, 319, 1346–1348. Available online: http://0-www-sciencedirect-com.brum.beds.ac.uk/science/article/pii/S0140673682924114 (accessed on 27 September 2015). [CrossRef]
- Gieseker, K.E. Outbreak of aflatoxin poisoning-Estern and Central Provinces, Kenya, January–July 2004. Public Health Fac. Publ. 2004, 53, 790–793. [Google Scholar]
- Cotty, P.J.; Antilla, L.; Wakelyn, P.J. Competitive exclusion of aflatoxin producers: Farmer-driven research and development. In Biological Control: A Global Perspective; CABI: Oxfordshire, UK, 2007; pp. 241–253. [Google Scholar]
- Probst, C.; Callicott, K.A.; Cotty, P.J. Deadly strains of Kenyan Aspergillus are distinct from other aflatoxin producers. Eur. J. Plant Pathol. 2012, 132, 419–429. [Google Scholar] [CrossRef]
- Lewis, L.; Onsongo, M.; Njapau, H.; Schurz-Rogers, H.; Luber, G.; Kieszak, S.; Nyamongo, J.; Backer, L.; Dahiye, A.M.; Misore, A.; et al. Aflatoxin Contamination of Commercial Maize Products during an Outbreak of Acute Aflatoxicosis in Eastern and Central Kenya. Environ. Health Perspect. 2005, 113, 1763–1767. Available online: http://www.ehponline.org/ambra-doi-resolver/10.1289/ehp.7998 (accessed on 15 May 2018). [CrossRef] [PubMed] [Green Version]
- Probst, C.; Schulthess, F.; Cotty, P.J. Impact of Aspergillus section Flavi community structure on the development of lethal levels of aflatoxins in Kenyan maize (Zea mays). J. Appl. Microbiol. 2010, 108, 600–610. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/19674186 (accessed on 15 May 2018). [CrossRef] [PubMed]
- De Coninck, B.M.A.; Amand, O.; Delauré, S.L.; Lucas, S.; Hias, N.; Weyens, G.; Mathys, J.; De Bruyne, E.; Cammue, B.P.A. The use of digital image analysis and real-time PCR fine-tunes bioassays for quantification of Cercospora leaf spot disease in sugar beet breeding. Plant Pathol. 2012, 61, 76–84. [Google Scholar] [CrossRef]
- Mackay, I.M. Real-Time PCR in Microbiology From Diagnosis to Characterization; Horizon Scientific Press: Poole, UK, 2007. [Google Scholar]
- Sanzani, S.M.; Li Destri Nicosia, M.G.; Faedda, R.; Cacciola, S.O.; Schena, L. Use of quantitative PCR detection methods to study biocontrol agents and phytopathogenic fungi and oomycetes in environmental samples. J. Phytopathol. 2014, 162, 1–13. [Google Scholar] [CrossRef]
- Mideros, S.X.; Windham, G.L.; Williams, W.P.; Nelson, R.J. Aspergillus flavus Biomass in Maize Estimated by Quantitative Real-Time Polymerase Chain Reaction Is Strongly Correlated with Aflatoxin Concentration. Plant Dis. 2009, 93, 1163–1170. [Google Scholar] [CrossRef]
- Mylroie, J.E.; Ozkan, S.; Shivaji, R.; LWindham, G.; Alpe, M.N.; PaulWilliams, W. Identification and Quantification of a Toxigenic and Non-Toxigenic Aspergillus flavus Strain in Contaminated Maize Using Quantitative Real-Time PCR. Toxins 2016, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Cotty, P.J. Biocompetitive exclusion of toxigenic fungi. In the Mycotoxin Factbook; Wageningen Academic Publishers: Wageningen, The Netherlands, 2006; pp. 179–197. [Google Scholar]
- Mehl, H.L.; Cotty, P.J. Variability in competitive ability among Aspergillus flavus vegetative compatibility groups during maize infection. Phytopathology 2008, 98, S103. [Google Scholar]
- Atehnkeng, J.; Donner, M.; Ojiambo, P.S.; Ikotun, B.; Augusto, J.; Cotty, P.J.; Bandyopadhyay, R. Environmental distribution and genetic diversity of vegetative compatibility groups determine biocontrol strategies to mitigate aflatoxin contamination of maize by Aspergillus flavus. Microb. Biotechnol. 2016, 9, 75–88. [Google Scholar] [CrossRef]
- Bandyopadhyay, R.; Ortega-Beltran, A.; Akande, A.; Mutegi, C.; Atehnkeng, J.; Kaptoge, L.; Senghor, A.L.; Adhikari, B.N.; Cotty, P.J. Biological control of aflatoxins in Africa: Current status and potential challenges in the face of climate change. World Mycotoxin J. 2016, 9, 771–789. [Google Scholar] [CrossRef]
- Cotty, P.J.; Jaime-Garcia, R. Influences of climate on aflatoxin producing fungi and aflatoxin contamination. Int. J. Food Microbiol. 2007, 119, 109–115. Available online: http://0-www-sciencedirect-com.brum.beds.ac.uk/science/article/pii/S0168160507003959 (accessed on 17 April 2016). [CrossRef] [PubMed]
- Payne, G.A.; Yu, J. Ecology, development and gene regulation in Aspergillus flavus. In Molecular Biology and Genomics; Caister Academic Press: Poole, UK, 2010. [Google Scholar]
- Wu, F.; Khlangwiset, P. Health economic impacts and cost-effectiveness of aflatoxin reduction strategies in Africa: Case studies in biocontrol and postharvest interventions. Food Addit. Contam. Part A 2010, 27, 496–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacoo, A.P.; Wendiro, D.; Vuzi, P.C.; Hawumba, J.F. Methods for Detection of Aflatoxins in Agricultural Food Crops. J. Appl. Chem. 2014, 1–15. Available online: http://www.hindawi.com/journals/jac/2014/706291/ (accessed on 18 May 2018). [CrossRef]
- Sulyok, M.; Krska, R.; Schuhmacher, R.; Sulyok, M.; Krska, R.; Schuhmacher, R. Application of a liquid chromatography—Tandem mass spectrometric method to multi-mycotoxin determination in raw cereals and evaluation of matrix effects. Food Addit. Contam. 2007, 10, 1184–1195. [Google Scholar] [CrossRef]
- Hayat, A.; Paniel, N.; Rhouati, A.; Marty, J.L.; Barthelmebs, L. Recent advances in ochratoxin A-producing fungi detection based on PCR methods and ochratoxin A analysis in food matrices. Food Control 2012, 26, 401–415. [Google Scholar] [CrossRef]
- Wei, D.; Zhou, L.; Selvaraj, J.N.; Zhang, C.; Xing, F.; Zhao, Y.; Wang, Y.; Liu, Y. Molecular characterization of atoxigenic Aspergillus flavus isolates collected in China. J. Microbiol. 2014, 52, 559–565. Available online: http://0-link-springer-com.brum.beds.ac.uk/10.1007/s12275-014-3629-8 (accessed on 18 May 2018). [CrossRef]
- Okoth, S.; De Boevre, M.; Vidal, A.; Diana Di Mavungu, J.; Landschoot, S.; Kyallo, M.; Njuguna, J.; Harvey, J.; De Saeger, S. Genetic and Toxigenic Variability within Aspergillus flavus Population Isolated from Maize in Two Diverse Environments in Kenya. Front. Microbiol. 2018, 9, 1–14. Available online: http://journal.frontiersin.org/article/10.3389/fmicb.2018.00057/full (accessed on 18 May 2018). [CrossRef] [PubMed]
- Blanco-meneses, M.; Ristaino, J.B.; Pathology, P.; Carolina, N.; Rica, C.; Carolina, N. Detection and Quantification of Peronospora tabacina Using a Real-Time Polymerase Chain Reaction Assay. Plant Dis. 2011, 95, 673–682. [Google Scholar] [CrossRef]
- Boutigny, A.L.; Beukes, I.; Small, I.; Zühlke, S.; Spiteller, M.; Van Rensburg, B.J.; Flett, B.; Viljoen, A. Quantitative detection of Fusarium pathogens and their mycotoxins in South African maize. Plant Pathol. 2012, 61, 522–531. [Google Scholar] [CrossRef]
- Songsermsakul, P.E.R.-F. A Review of Recent Trends in Applications of Liquid Chromatography-Mass Spectrometry for Determination of Mycotoxins Liquid Chromatography-Mass Spectrometry for determination of mycotoxins. J. Liq. Chromatogr. Relat. Technol. 2008, 6076, 1641–1686. [Google Scholar] [CrossRef]
- Malachová, A.; Sulyok, M.; Beltrán, E.; Berthiller, F.; Krska, R. Optimization and validation of a quantitative liquid chromatography-tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J. Chromatogr. A 2014, 1362, 145–156. [Google Scholar] [CrossRef]
- Mitema, A.; Okoth, S.; Rafudeen, M.S. Vegetative compatibility and phenotypic characterization as a means of determining genetic diversity of Aspergillus fl avus isolates. Fungal Biol. 2018, 122, 203–213. [Google Scholar] [CrossRef]
- Zöllner, P.; Mayer-Helm, B. Trace mycotoxin analysis in complex biological and food matrices by liquid chromatography-atmospheric pressure ionisation mass spectrometry. J. Chromatogr. A 2006, 1136, 123–169. [Google Scholar] [CrossRef]
- Azziz-baumgartner, E.; Lindblade, K.; Gieseker, K.; Rogers, H.S.; Kieszak, S.; Njapau, H.; Schleicher, R.; McCoy, L.F.; Misore, A.; DeCock, K.; et al. Case—Control Study of an Acute Aflatoxicosis Outbreak, Kenya, 2004. Environ Health Perspect. 2005, 113, 1779–1783. [Google Scholar] [CrossRef]
- Debode, J.; Van Hemelrijck, W.; Baeyen, S.; Creemers, P.; Heungens, K.; Maes, M. Quantitative detection and monitoring of Colletotrichum acutatum in strawberry leaves using real-time PCR. Plant Pathol. 2009, 58, 504–514. [Google Scholar] [CrossRef]
- Divon, H.H.; Razzaghian, J. Fusarium langsethiae (Torp and Nirenberg), investigation of alternative infection routes in oats. Eur. J. Plant Pathol. 2012, 132, 147–161. [Google Scholar] [CrossRef]
- Gallo, A.; Masoero, F.; Bertuzzi, T.; Piva, G.; Pietri, A. Effect of the inclusion of adsorbents on aflatoxin B-1 quantification in animal feedstuffs. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2010, 27, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Horn, B.W.; Greene, R.L. Vegetative compatibility within populations of Aspergillus flavus, A. parasiticus, and A. tamarii from a peanut field. Mycologia 1995, 87, 324–332. [Google Scholar] [CrossRef]
- Dorner, J.W. Biological Control of Aflatoxin Contamination of Crops. Toxin Rev. 2004, 23, 425–450. [Google Scholar] [CrossRef]
- Hruska, Z.; Rajasekaran, K.; Yao, H.; Kinkaid, R.; Darlington, D.; Brown, R.L.; Bhatnagar, D.; Cleveland, T.E. Co-inoculation of aflatoxigenic and non-aflatoxigenic strains of Aspergillus flavus to study fungal invasion, colonization, and competition in maize kernels. Front. Microbiol. 2014, 5, 122. Available online: http://0-www-scopus-com.brum.beds.ac.uk/inward/record.url?eid=2-s2.0-84897941698&partnerID=tZOtx3y1 (accessed on 18 May 2018). [CrossRef] [PubMed]
- Huang, C.; Jha, A.; Sweany, R.; DeRobertis, C.; Damann, K.E. Intraspecific aflatoxin inhibition in Aspergillus flavus is thigmoregulated, independent of vegetative compatibility group and is strain dependent. PLoS ONE 2011, 6, e23470. Available online: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3158758&tool=pmcentrez&rendertype=abstract (accessed on 18 May 2018). [CrossRef] [PubMed]
- Mehl, H.L.; Cotty, P.J. Influence of plant host species on intraspecific competition during infection by Aspergillus flavus. Plant Pathol. 2013, 62, 1310–1318. [Google Scholar] [CrossRef]
- Mitema, A.O. Molecular and Bio-Analytical Characterisation as a Means to Understand Genetic Diversity within Kenyan Aspergillus flavus Strains. Doctoral Dissertation, University of Cape Town, Cape Town, South Africa, 2018. [Google Scholar]
- Mehl, H.L.; Jaime, R.; Callicott, K.A.; Probst, C.; Garber, N.P.; Ortega-Beltran, A.; Grubisha, L.C.; Cotty, P.J. Aspergillus flavus diversity on crops and in the environment can be exploited to reduce aflatoxin exposure and improve health. Ann. N. Y. Acad. Sci. 2012, 1273, 7–17. [Google Scholar] [CrossRef]
- Medina, A.; Schmidt-Heydt, M.; Rodriguez, A.; Parra, R.; Geisen, R.; Magan, N. Impacts of environmental stress on growth, secondary metabolite biosynthetic gene clusters and metabolite production of xerotolerant/xerophilic fungi. Curr. Genet. 2015, 61, 325–334. [Google Scholar] [CrossRef]
- De Lima Alves, F.; Stevenson, A.; Baxter, E.; Gillion, J.L.; Hejazi, F.; Hayes, S.; Morrison, I.E.; Prior, B.A.; McGenity, T.J.; Rangel, D.E.; et al. Concomitant osmotic and chaotropicity—Induced stresses in Aspergillus wentii: Compatible solutes determine the biotic window. Curr. Genet. 2015, 61, 457–477. [Google Scholar] [CrossRef] [PubMed]
- Sunda, W.; Ochuodho, J.; Ngode, L.; Okalebo, J.R.; Othieno, C.O.; Nekesa, A.O.; Kipkoech, A.K. Development of integrated Striga management package to improve maize production in Western Kenya. In Proceedings of the Third RUFORUM Biennial Meeting, Entebbe, Uganda, 24–28 September 2012; pp. 375–381. [Google Scholar]
- Möller, E.M.; Bahnweg, G.; Sandermann, H.; Geiger, H.H. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res. 1992, 20, 6115–6116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Manoli, A.; Sturaro, A.; Trevisan, S.; Quaggiotti, S.; Nonis, A. Evaluation of candidate reference genes for qPCR in maize. J. Plant Physiol. 2012, 169, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Nicolaisen, M.; Kærgaard, L.; Lazzaro, I.; Henrik, N.; Fejer, A. Real-time PCR for quanti fi cation of eleven individual Fusarium species in cereals. J. Microbiol. Methods 2009, 76, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonák, J.; Lind, K.; Sindelka, R.; Sjöback, R.; Sjögreen, B.; Strömbom, L.; et al. The real-time polymerase chain reaction. Mol. Asp. Med. 2006, 27, 95–125. [Google Scholar] [CrossRef] [PubMed]
- Sardiñas, N.; Vázquez, C.; Gil-serna, J.; González-jaén, M.T.; Patiño, B. International Journal of Food Microbiology Speci fi c detection and quanti fi cation of Aspergillus fl avus and Aspergillus parasiticus in wheat fl our by SYBR® Green quantitative PCR. Int. J. Food Microbiol. 2011, 145, 121–125. [Google Scholar] [CrossRef]
- Mehl, H.L.; Cotty, P.J. Variation in Competitive Ability Among Isolates of Aspergillus flavus from Different Vegetative Compatibility Groups During Maize Infection. Mycology 2010, 100, 151. [Google Scholar]
- Abbas, H.K.; Zablotowicz, R.M.; Bruns, H.A.; Abel, C.A. Biocontrol of aflatoxin in corn by inoculation with non-aflatoxigenic Aspergillus flavus isolates. Biocontrol Sci. Technol. 2006, 16, 437–449. Available online: http://0-www-tandfonline-com.brum.beds.ac.uk/doi/abs/10.1080/09583150500532477 (accessed on 18 May 2018). [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotypic Characteristic | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Maize Line | Control Roots | Control Shoots | Infected Roots | Infected Shoots | ||||||||||||
| Exp 1 | Exp 2 | Exp 3 | Average (mm) | Exp 1 | Exp 2 | Exp 3 | Average (mm) | Exp 1 | Exp 2 | Exp 3 | Average (mm) | Exp 1 | Exp 2 | Exp 3 | Average (mm) | |
| GAF4 | 285 | 260 | 278 | 274.33 | 352 | 322 | 312 | 328.67 | 134 | 113 | 98 | 115 | 142 | 185 | 111 | 146 |
| KDV1 | 272 | 252 | 232 | 252 | 344 | 300 | 323 | 322.33 | 78 | 83 | 84 | 81.67 | 82 | 87 | 91 | 86.67 |
| Primer Name | Forward Primer (5′-3′) | Reverse Primer (5′-3′) | Product Size (bp) | Ta | Reference |
|---|---|---|---|---|---|
| Membrane Protein (MEP) | TGTACTCGGCAATGCTCTTG | TTTGATGCTCCAGGCTTACC | 203 | 64 °C | Manoli et al. [51] |
| Elongation Factor 1 alpha (EF1α) | CGTTTCTGCCCTCTCCCA | TGCTTGACACGTGACGATGA | 102 | 62 °C | Nicolaisen et al. [52] |
| β-TubulinM | TCTTCATGGTTGGCTTCGCT | CTTGGGTCGAACATCTGCT | 118 | 62 °C | Mitema et al. [33] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitema, A.; Okoth, S.; Rafudeen, S.M. The Development of a qPCR Assay to Measure Aspergillus flavus Biomass in Maize and the Use of a Biocontrol Strategy to Limit Aflatoxin Production. Toxins 2019, 11, 179. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030179
Mitema A, Okoth S, Rafudeen SM. The Development of a qPCR Assay to Measure Aspergillus flavus Biomass in Maize and the Use of a Biocontrol Strategy to Limit Aflatoxin Production. Toxins. 2019; 11(3):179. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030179
Chicago/Turabian StyleMitema, Alfred, Sheila Okoth, and Suhail M. Rafudeen. 2019. "The Development of a qPCR Assay to Measure Aspergillus flavus Biomass in Maize and the Use of a Biocontrol Strategy to Limit Aflatoxin Production" Toxins 11, no. 3: 179. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030179