Intracellular Transport and Cytotoxicity of the Protein Toxin Ricin

 , , , and
, , , and 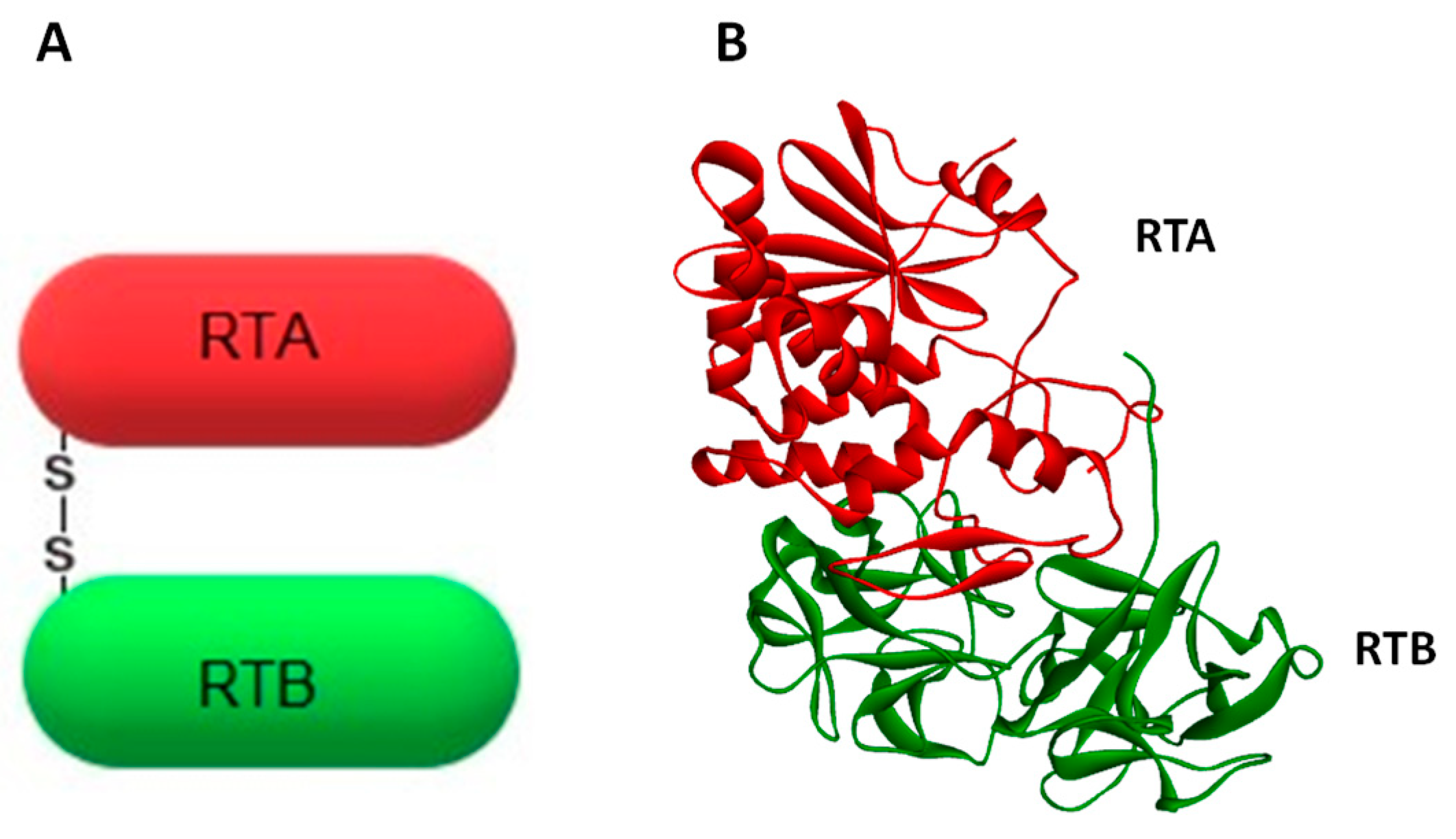{kind=link}
{kind=link}
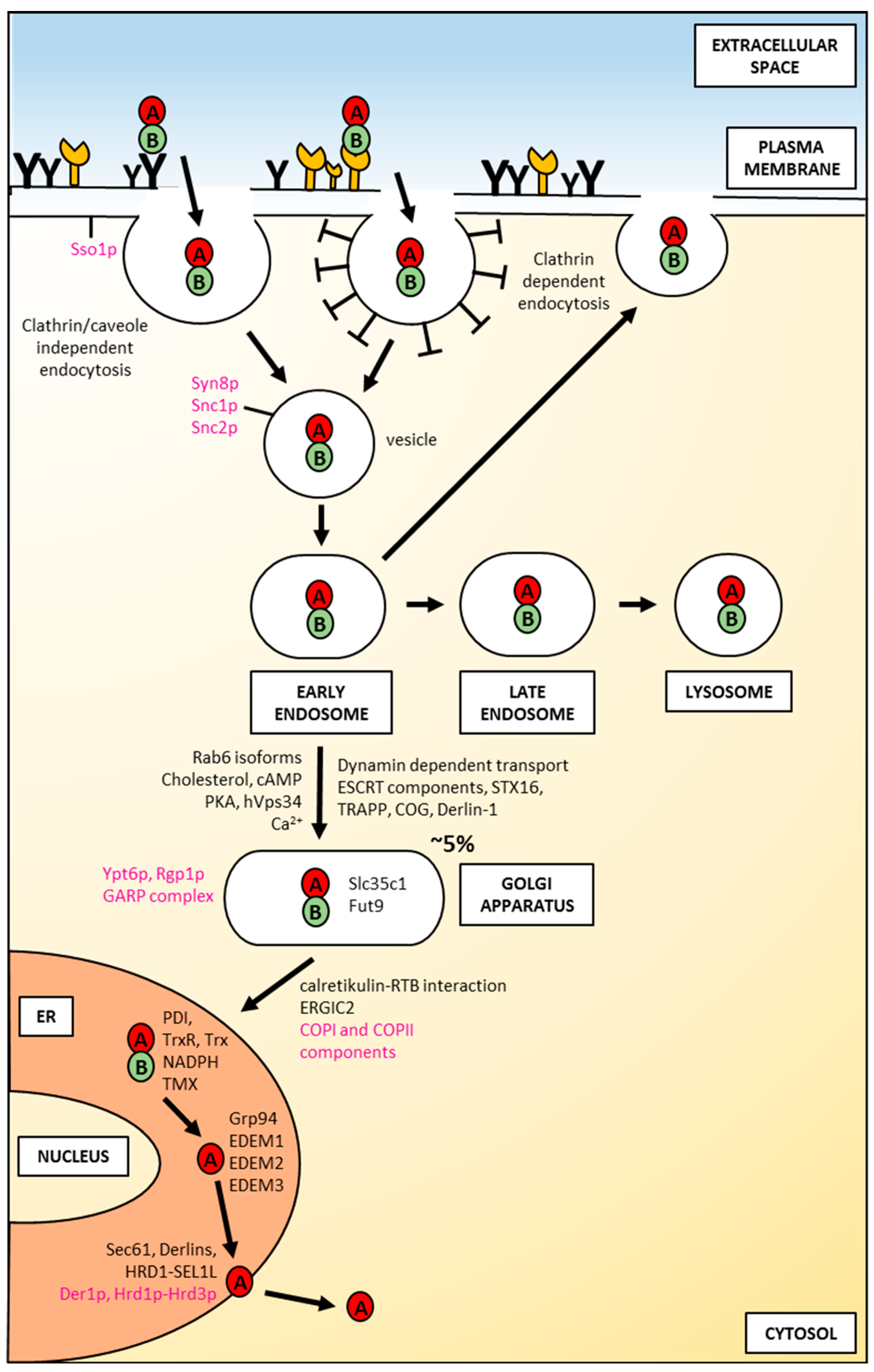{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Intracellular Transport of Ricin
2.1. Uptake of Ricin into the Cell
2.2. Intracellular Routing of Ricin to the Endoplasmic Reticulum
2.3. Ricin Translocation to the Cytosol
3. Cytotoxic Action of Ricin on Cells
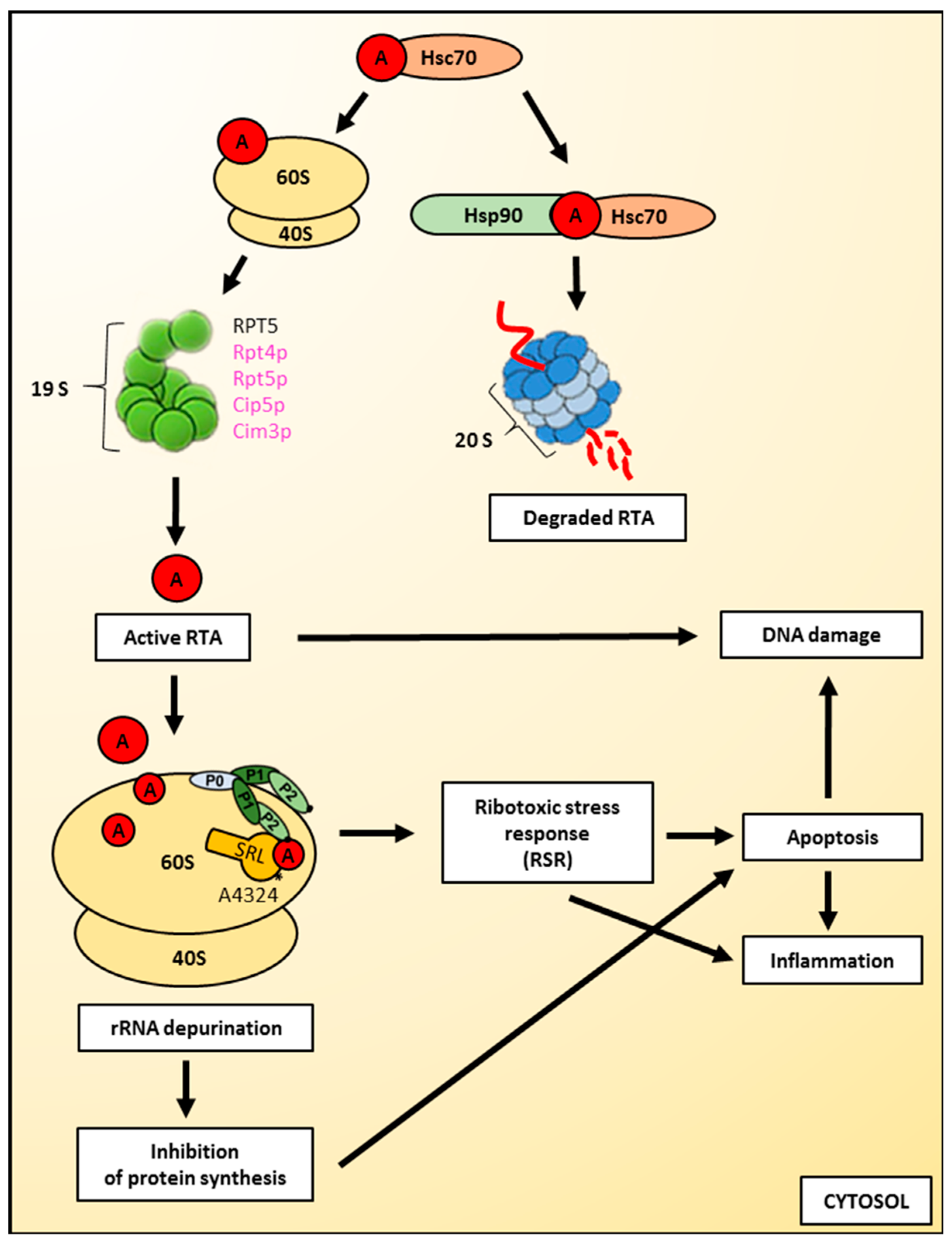
3.1. Activation of Ricin A-Chain in the Cytosol Regulation of RTA Folding versus Degradation
3.2. Ricin A-Chain Action on Ribosomes
3.3. Mechanisms of Ricin-Induced Apoptosis
3.3.1. Ricin-Induced Activation of Caspases
3.3.2. Activation of Bcl-2 Family Members by Ricin
3.3.3. Activation of Stress Associated Signaling Pathways by Ricin
3.3.4. Direct Action of Ricin on DNA and Ricin-Mediated Inhibition of DNA Repair Enzymes
3.3.5. Ricin-Mediated Reactive Oxygen Species Production
3.3.6. Ricin B-Chain-Induced Apoptosis
4. Perspectives
4.1. Ricin-Based Immunotoxins
4.2. Ricin Conjugated with Nanoparticles
4.3. Vaccines against Ricin and Neutralizing Antibodies against Ricin
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Stillmark, H. Ueber Ricin, ein giftiges Ferment aus den Samen von Ricinus comm. L. und einigen anderen Euphorbiaceen: Inaugural-Dissertation. MD Thesis, University of Dorpat, Dorpat, Estonia, 1888. [Google Scholar]
- Boyd, W.C.; Shapleigh, E. Diagnosis by subgroups of blood groups A and AB by use of plant agglutinins (lectins). J. Lab. Clin. Med. 1954, 44, 235–237. [Google Scholar] [PubMed]
- Olsnes, S.; Pihl, A. Ricin—A potent inhibitor of protein synthesis. FEBS Lett. 1972, 20, 327–329. [Google Scholar] [CrossRef]
- Olsnes, S.; Pihl, A. Different biological properties of the two constituent peptide chains of ricin, a toxic protein inhibiting protein synthesis. Biochemistry 1973, 12, 3121–3126. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, S.; Heiberg, R.; Pihl, A. Inactivation of eucaryotic ribosomes by the toxic plant proteins abrin and ricin. Mol. Biol. Rep. 1973, 1, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar]
- Lord, J.M.; Roberts, L.M.; Robertus, J.D. Ricin: Structure, mode of action, and some current applications. FASEB J. 1994, 8, 201–208. [Google Scholar] [CrossRef]
- Olsnes, S.; Refsnes, K.; Pihl, A. Mechanism of action of the toxic lectins abrin and ricin. Nature 1974, 249, 627–631. [Google Scholar] [CrossRef]
- Shi, W.-W.; Mak, A.N.-S.; Wong, K.-B.; Shaw, P.-C. Structures and Ribosomal Interaction of Ribosome-Inactivating Proteins. Molecules 2016, 21, 1588. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.-P.; Kahn, J.N.; Tumer, N.E. Functional Assays for Measuring the Catalytic Activity of Ribosome Inactivating Proteins. Toxins 2018, 10, 240. [Google Scholar] [CrossRef]
- Barbieri, L.; Battelli, M.G.; Stirpe, F. Ribosome-inactivating proteins from plants. Biochim. Biophys. Acta 1993, 1154, 237–282. [Google Scholar] [CrossRef]
- Voorhees, R.M.; Schmeing, T.M.; Kelley, A.C.; Ramakrishnan, V. The mechanism for activation of GTP hydrolysis on the ribosome. Science 2010, 330, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Khade, P.K.; Sanbonmatsu, K.Y.; Joseph, S. Functional role of the sarcin-ricin loop of the 23S rRNA in the elongation cycle of protein synthesis. J. Mol. Biol. 2012, 419, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, L.; Lambert, N.J.; Maklan, E.J.; Horan, L.H.; Noller, H.F. The sarcin–ricin loop of 23S rRNA is essential for assembly of the functional core of the 50S ribosomal subunit. RNA 2008, 14, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Moazed, D.; Robertson, J.M.; Noller, H.F. Interaction of elongation factors EF-G and EF-Tu with a conserved loop in 23S RNA. Nature 1988, 334, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Hartley, M.R.; Lord, J.M. Cytotoxic ribosome-inactivating lectins from plants. Biochim. Biophys. Acta 2004, 1701, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, S.; Fernandez-Puentes, C.; Carrasco, L.; Vazquez, D. Ribosome inactivation by the toxic lectins abrin and ricin. Kinetics of the enzymic activity of the toxin A-chains. Eur. J. Biochem. 1975, 60, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, S. The history of ricin, abrin and related toxins. Toxicon 2004, 44, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Eiklid, K.; Olsnes, S.; Pihl, A. Entry of lethal doses of abrin, ricin and modeccin into the cytosol of HeLa cells. Exp. Cell Res. 1980, 126, 321–326. [Google Scholar] [CrossRef]
- Olson, K.R. Poisoning and Drug Overdose, Sixth Edition, 6th ed.; McGraw-Hill Education/Medical: New York, NY, USA, 2011; ISBN 978-0-07-166833-0. [Google Scholar]
- Moshiri, M.; Hamid, F.; Etemad, L. Ricin Toxicity: Clinical and Molecular Aspects. Rep. Biochem. Mol. Biol. 2016, 4, 60–65. [Google Scholar]
- Convention on the Prohibition of the Development, Production, Stockpiling and Use of Chemical Weapons and on Their Destruction; Organisation for the Probhibition of Chemical Weapons (OPCW): The Hague, The Netherlands, 2005.
- Li, X.-P.; Baricevic, M.; Saidasan, H.; Tumer, N.E. Ribosome depurination is not sufficient for ricin-mediated cell death in Saccharomyces cerevisiae. Infect. Immun. 2007, 75, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Flexner, S. The histological changes produced by ricin and abrin intoxications. J. Exp. Med. 1897, 2, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.D.; Leek, M.D.; Gee, D.J. The toxic plant proteins ricin and abrin induce apoptotic changes in mammalian lymphoid tissues and intestine. J. Pathol. 1987, 151, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.N.; Lindsay, C.D.; Griffiths, G.D. Morphology of ricin and abrin exposed endothelial cells is consistent with apoptotic cell death. Hum. Exp. Toxicol. 1996, 15, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Day, P.J.; Pinheiro, T.J.T.; Roberts, L.M.; Lord, J.M. Binding of ricin A-chain to negatively charged phospholipid vesicles leads to protein structural changes and destabilizes the lipid bilayer. Biochemistry 2002, 41, 2836–2843. [Google Scholar] [CrossRef] [PubMed]
- Kumar, O.; Sugendran, K.; Vijayaraghavan, R. Oxidative stress associated hepatic and renal toxicity induced by ricin in mice. Toxicon 2003, 41, 333–338. [Google Scholar] [CrossRef]
- Słomińska-Wojewódzka, M.; Sandvig, K. Ricin and Ricin-Containing Immunotoxins: Insights into Intracellular Transport and Mechanism of action in Vitro. Antibodies 2013, 2, 236–269. [Google Scholar] [CrossRef] [Green Version]
- Shih, S.-F.; Wu, Y.-H.; Hung, C.-H.; Yang, H.-Y.; Lin, J.-Y. Abrin Triggers Cell Death by Inactivating a Thiol-specific Antioxidant Protein. J. Biol. Chem. 2001, 276, 21870–21877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluna, R.; Coleman, E.; Jones, C.; Ghetie, V.; Vitetta, E.S. The Effect of a Monoclonal Antibody Coupled to Ricin A Chain-Derived Peptides on Endothelial Cells in Vitro: Insights into Toxin-Mediated Vascular Damage. Exp. Cell Res. 2000, 258, 417–424. [Google Scholar] [CrossRef]
- Hasegawa, N.; Kimura, Y.; Oda, T.; Komatsu, N.; Muramatsu, T.H.-J. Isolated ricin B-chain-mediated apoptosis in U937 cells. Biosci. Biotechnol. Biochem. 2000, 64, 1422–1429. [Google Scholar] [CrossRef]
- Berger, T.; Eisenkraft, A.; Bar-Haim, E.; Kassirer, M.; Aran, A.A.; Fogel, I. Toxins as biological weapons for terror-characteristics, challenges and medical countermeasures: A mini-review. Disaster Mil. Med. 2016, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Janik, E.; Ceremuga, M.; Saluk-Bijak, J.; Bijak, M. Biological Toxins as the Potential Tools for Bioterrorism. Int. J. Mol. Sci. 2019, 20, 1181. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Yin, J.; Chau, D.; Hu, C.C.; Cherwonogrodzky, J.W. Anti-Ricin Protective Monoclonal Antibodies. Ricin Toxin 2014, 14, 145–158. [Google Scholar]
- Herrera, C.; Klokk, T.I.; Cole, R.; Sandvig, K.; Mantis, N.J. A Bispecific Antibody Promotes Aggregation of Ricin Toxin on Cell Surfaces and Alters Dynamics of Toxin Internalization and Trafficking. PLoS ONE 2016, 11, e0156893. [Google Scholar] [CrossRef]
- Yermakova, A.; Klokk, T.I.; O’Hara, J.M.; Cole, R.; Sandvig, K.; Mantis, N.J. Neutralizing Monoclonal Antibodies against Disparate Epitopes on Ricin Toxin’s Enzymatic Subunit Interfere with Intracellular Toxin Transport. Sci. Rep. 2016, 6, 22721. [Google Scholar] [CrossRef] [PubMed]
- Brey, R.N.; Mantis, N.J.; Pincus, S.H.; Vitetta, E.S.; Smith, L.A.; Roy, C.J. Recent advances in the development of vaccines against ricin. Hum. Vaccin Immunother. 2016, 12, 1196–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Pop, L.M.; Schindler, J.; Vitetta, E.S. Immunotoxins constructed with chimeric, short-lived anti-CD22 monoclonal antibodies induce less vascular leak without loss of cytotoxicity. MAbs 2012, 4, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iversen, T.G.; Frerker, N.; Sandvig, K. Uptake of ricinB-quantum dot nanoparticles by a macropinocytosis-like mechanism. J. Nanobiotechnol. 2012, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Tyagi, M.; Pachauri, M.; Ghosh, P.C. Potential therapeutic applications of plant toxin-ricin in cancer: Challenges and advances. Tumor Biol. 2015, 36, 8239–8246. [Google Scholar] [CrossRef]
- Pizzo, E.; Di Maro, A. A new age for biomedical applications of Ribosome Inactivating Proteins (RIPs): From bioconjugate to nanoconstructs. J. Biomed Sci. 2016, 23, 54. [Google Scholar] [CrossRef]
- Jiao, P.; Zhang, J.; Dong, Y.; Wei, D.; Ren, Y. Construction and characterization of the recombinant immunotoxin RTA-4D5-KDEL targeting HER2/neu-positive cancer cells and locating the endoplasmic reticulum. Appl. Microbiol. Biotechnol. 2018, 102, 9585–9594. [Google Scholar] [CrossRef]
- Magnusson, S.; Kjeken, R.; Berg, T. Characterization of two distinct pathways of endocytosis of ricin by rat liver endothelial cells. Exp. Cell Res. 1993, 205, 118–125. [Google Scholar] [CrossRef]
- Sphyris, N.; Lord, J.M.; Wales, R.; Roberts, L.M. Mutational analysis of the Ricinus lectin B-chains. Galactose-binding ability of the 2 gamma subdomain of Ricinus communis agglutinin B-chain. J. Biol. Chem. 1995, 270, 20292–20297. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Olsnes, S.; Pihl, A. Kinetics of binding of the toxic lectins abrin and ricin to surface receptors of human cells. J. Biol. Chem. 1976, 251, 3977–3984. [Google Scholar]
- Sandvig, K.; Spilsberg, B.; Lauvrak, S.U.; Torgersen, M.L.; Iversen, T.-G.; van Deurs, B.O. Pathways followed by protein toxins into cells. Int. J. Med. Microbiol. 2004, 293, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Van Deurs, B.; Pedersen, L.R.; Sundan, A.; Olsnes, S.; Sandvig, K. Receptor-mediated endocytosis of a ricin-colloidal gold conjugate in vero cells. Intracellular routing to vacuolar and tubulo-vesicular portions of the endosomal system. Exp. Cell Res. 1985, 159, 287–304. [Google Scholar] [CrossRef]
- Moya, M.; Dautry-Varsat, A.; Goud, B.; Louvard, D.; Boquet, P. Inhibition of coated pit formation in Hep2 cells blocks the cytotoxicity of diphtheria toxin but not that of ricin toxin. J. Cell Biol. 1985, 101, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Olsnes, S.; Petersen, O.W.; van Deurs, B. Inhibition of endocytosis from coated pits by acidification of the cytosol. J. Cell. Biochem. 1988, 36, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.C.; Smith, D.C.; Roberts, L.M.; Lord, J.M. Expression of mutant dynamin protects cells against diphtheria toxin but not against ricin. Exp. Cell Res. 1998, 239, 293–300. [Google Scholar] [CrossRef]
- Spooner, R.A.; Lord, J.M. Ricin Trafficking in Cells. Toxins 2015, 7, 49–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodal, S.K.; Skretting, G.; Garred, O.; Vilhardt, F.; van Deurs, B.; Sandvig, K. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol. Biol. Cell 1999, 10, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Pust, S.; Skotland, T.; van Deurs, B. Clathrin-independent endocytosis: Mechanisms and function. Curr. Opin. Cell Biol. 2011, 23, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Kavaliauskiene, S.; Skotland, T. Clathrin-independent endocytosis: An increasing degree of complexity. Histochem. Cell Biol. 2018, 150, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Grimmer, S.; Iversen, T.G.; Rodal, K.; Torgersen, M.L.; Nicoziani, P.; van Deurs, B. Ricin transport into cells: Studies of endocytosis and intracellular transport. Int. J. Med. Microbiol. 2000, 290, 415–420. [Google Scholar] [CrossRef]
- Sokołowska, I.; Wälchli, S.; Węgrzyn, G.; Sandvig, K.; Słomińska-Wojewódzka, M. A single point mutation in ricin A-chain increases toxin degradation and inhibits EDEM1-dependent ER retrotranslocation. Biochem. J. 2011, 436, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Sokołowska, I.; Piłka, E.S.; Sandvig, K.; Węgrzyn, G.; Słomińska-Wojewódzka, M. Hydrophobicity of protein determinants influences the recognition of substrates by EDEM1 and EDEM2 in human cells. BMC Cell Biol. 2015, 16, 1. [Google Scholar] [CrossRef]
- Becker, B.; Schnöder, T.; Schmitt, M.J. Yeast Reporter Assay to Identify Cellular Components of Ricin Toxin A Chain Trafficking. Toxins 2016, 8, 366. [Google Scholar] [CrossRef]
- Lewis, M.J.; Pelham, H.R.B. A new yeast endosomal SNARE related to mammalian syntaxin 8. Traffic 2002, 3, 922–929. [Google Scholar] [CrossRef]
- Tang, H.; Song, M.; He, Y.; Wang, J.; Wang, S.; Shen, Y.; Hou, J.; Bao, X. Engineering vesicle trafficking improves the extracellular activity and surface display efficiency of cellulases in Saccharomyces cerevisiae. Biotechnol. Biofuels 2017, 10, 53. [Google Scholar] [CrossRef]
- Sandvig, K.; Olsnes, S.; Pihl, A. Interactions between abrus lectins and Sephadex particles possessing immobilized desialylated fetuin. Model studies of the interaction of lectins with cell surface receptors. Eur. J. Biochem. 1978, 88, 307–313. [Google Scholar] [CrossRef]
- Blum, J.S.; Fiani, M.L.; Stahl, P.D. Proteolytic cleavage of ricin A chain in endosomal vesicles. Evidence for the action of endosomal proteases at both neutral and acidic pH. J. Biol. Chem. 1991, 266, 22091–22095. [Google Scholar] [PubMed]
- Sandvig, K.; van Deurs, B. Membrane traffic exploited by protein toxins. Annu. Rev. Cell Dev. Biol. 2002, 18, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Van Deurs, B.; Tønnessen, T.I.; Petersen, O.W.; Sandvig, K.; Olsnes, S. Routing of internalized ricin and ricin conjugates to the Golgi complex. J. Cell Biol. 1986, 102, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Van Deurs, B.; Sandvig, K.; Petersen, O.W.; Olsnes, S.; Simons, K.; Griffiths, G. Estimation of the amount of internalized ricin that reaches the trans-Golgi network. J. Cell Biol. 1988, 106, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; van Deurs, B. Delivery into cells: Lessons learned from plant and bacterial toxins. Gene Ther. 2005, 12, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Rapak, A.; Falnes, P.O.; Olsnes, S. Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc. Natl. Acad. Sci. USA 1997, 94, 3783–3788. [Google Scholar] [CrossRef] [Green Version]
- Driouich, A.; Zhang, G.F.; Staehelin, L.A. Effect of brefeldin A on the structure of the Golgi apparatus and on the synthesis and secretion of proteins and polysaccharides in sycamore maple (Acer pseudoplatanus) suspension-cultured cells. Plant Physiol. 1993, 101, 1363–1373. [Google Scholar] [CrossRef]
- Yoshida, T.; Chen, C.C.; Zhang, M.S.; Wu, H.C. Disruption of the Golgi apparatus by brefeldin A inhibits the cytotoxicity of ricin, modeccin, and Pseudomonas toxin. Exp. Cell Res. 1991, 192, 389–395. [Google Scholar] [CrossRef]
- Sandvig, K.; Prydz, K.; Hansen, S.H.; van Deurs, B. Ricin transport in brefeldin A-treated cells: Correlation between Golgi structure and toxic effect. J. Cell Biol. 1991, 115, 971–981. [Google Scholar] [CrossRef]
- Prydz, K.; Hansen, S.H.; Sandvig, K.; van Deurs, B. Effects of brefeldin A on endocytosis, transcytosis and transport to the Golgi complex in polarized MDCK cells. J. Cell Biol. 1992, 119, 259–272. [Google Scholar] [CrossRef]
- Leitinger, B.; Brown, J.L.; Spiess, M. Tagging secretory and membrane proteins with a tyrosine sulfation site. Tyrosine sulfation precedes galactosylation and sialylation in COS-7 cells. J. Biol. Chem. 1994, 269, 8115–8121. [Google Scholar]
- Llorente, A.; Rapak, A.; Schmid, S.L.; van Deurs, B.; Sandvig, K. Expression of Mutant Dynamin Inhibits Toxicity and Transport of Endocytosed Ricin to the Golgi Apparatus. J. Cell Biol. 1998, 140, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, J.L.; von Delft, F.; Brennan, P. Targeting the Small GTPase Superfamily through their Regulatory Proteins. Angew. Chem. Int. Ed. 2019. [Google Scholar] [CrossRef]
- Lombardi, D.; Soldati, T.; Riederer, M.A.; Goda, Y.; Zerial, M.; Pfeffer, S.R. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 1993, 12, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Riederer, M.A.; Soldati, T.; Shapiro, A.D.; Lin, J.; Pfeffer, S.R. Lysosome biogenesis requires Rab9 function and receptor recycling from endosomes to the trans-Golgi network. J. Cell Biol 1994, 125, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Iversen, T.G.; Skretting, G.; Llorente, A.; Nicoziani, P.; van Deurs, B.; Sandvig, K. Endosome to Golgi transport of ricin is independent of clathrin and of the Rab9- and Rab11-GTPases. Mol. Biol. Cell 2001, 12, 2099–2107. [Google Scholar] [CrossRef]
- Mallard, F.; Tang, B.L.; Galli, T.; Tenza, D.; Saint-Pol, A.; Yue, X.; Antony, C.; Hong, W.; Goud, B.; Johannes, L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J. Cell Biol. 2002, 156, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echard, A.; Opdam, F.J.; de Leeuw, H.J.; Jollivet, F.; Savelkoul, P.; Hendriks, W.; Voorberg, J.; Goud, B.; Fransen, J.A. Alternative splicing of the human Rab6A gene generates two close but functionally different isoforms. Mol. Biol. Cell 2000, 11, 3819–3833. [Google Scholar] [CrossRef]
- Mallard, F.; Antony, C.; Tenza, D.; Salamero, J.; Goud, B.; Johannes, L. Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of shiga toxin B-fragment transport. J. Cell Biol. 1998, 143, 973–990. [Google Scholar] [CrossRef]
- Utskarpen, A.; Slagsvold, H.H.; Iversen, T.-G.; Wälchli, S.; Sandvig, K. Transport of ricin from endosomes to the Golgi apparatus is regulated by Rab6A and Rab6A′. Traffic 2006, 7, 663–672. [Google Scholar] [CrossRef]
- Grimmer, S.; Iversen, T.-G.; van Deurs, B.; Sandvig, K. Endosome to Golgi Transport of Ricin Is Regulated by Cholesterol. Mol. Biol. Cell 2000, 11, 4205–4216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauvrak, S.U.; Llorente, A.; Iversen, T.-G.; Sandvig, K. Selective regulation of the Rab9-independent transport of ricin to the Golgi apparatus by calcium. J. Cell Sci. 2002, 115, 3449–3456. [Google Scholar] [PubMed]
- Porat, A.; Elazar, Z. Regulation of intra-Golgi membrane transport by calcium. J. Biol. Chem. 2000, 275, 29233–29237. [Google Scholar] [CrossRef] [PubMed]
- Moreau, D.; Kumar, P.; Wang, S.C.; Chaumet, A.; Chew, S.Y.; Chevalley, H.; Bard, F. Genome-wide RNAi screens identify genes required for Ricin and PE intoxications. Dev. Cell 2011, 21, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Schmitt, M.J. A Simple Fluorescence-based Reporter Assay to Identify Cellular Components Required for Ricin Toxin A Chain (RTA) Trafficking in Yeast. J. Vis. Exp. 2017, 130, e56588. [Google Scholar] [CrossRef] [PubMed]
- Birkeli, K.A.; Llorente, A.; Torgersen, M.L.; Keryer, G.; Taskén, K.; Sandvig, K. Endosome-to-Golgi transport is regulated by protein kinase A type II alpha. J. Biol. Chem. 2003, 278, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Skånland, S.S.; Wälchli, S.; Utskarpen, A.; Wandinger-Ness, A.; Sandvig, K. Phosphoinositide-regulated retrograde transport of ricin: Crosstalk between hVps34 and sorting nexins. Traffic 2007, 8, 297–309. [Google Scholar] [CrossRef]
- Sandvig, K.; van Deurs, B. Entry of ricin and Shiga toxin into cells: Molecular mechanisms and medical perspectives. EMBO J. 2000, 19, 5943–5950. [Google Scholar] [CrossRef]
- Day, P.J.; Owens, S.R.; Wesche, J.; Olsnes, S.; Roberts, L.M.; Lord, J.M. An interaction between ricin and calreticulin that may have implications for toxin trafficking. J. Biol. Chem. 2001, 276, 7202–7208. [Google Scholar] [CrossRef]
- Spooner, R.A.; Smith, D.C.; Easton, A.J.; Roberts, L.M.; Lord, J.M. Retrograde transport pathways utilised by viruses and protein toxins. Virol. J. 2006, 3, 26. [Google Scholar] [CrossRef]
- Chen, A.; AbuJarour, R.J.; Draper, R.K. Evidence that the transport of ricin to the cytoplasm is independent of both Rab6A and COPI. J. Cell Sci. 2003, 116, 3503–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorente, A.; Lauvrak, S.U.; van Deurs, B.; Sandvig, K. Induction of direct endosome to endoplasmic reticulum transport in Chinese hamster ovary (CHO) cells (LdlF) with a temperature-sensitive defect in epsilon-coatomer protein (epsilon-COP). J. Biol. Chem. 2003, 278, 35850–35855. [Google Scholar] [CrossRef] [PubMed]
- Otte, S.; Belden, W.J.; Heidtman, M.; Liu, J.; Jensen, O.N.; Barlowe, C. Erv41p and Erv46p: New components of COPII vesicles involved in transport between the ER and Golgi complex. J. Cell Biol. 2001, 152, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Adolf, F.; Rhiel, M.; Hessling, B.; Gao, Q.; Hellwig, A.; Béthune, J.; Wieland, F.T. Proteomic Profiling of Mammalian COPII and COPI Vesicles. Cell Rep. 2019, 26, 250–265. [Google Scholar] [CrossRef]
- Sato, K.; Sato, M.; Nakano, A. Rer1p, a Retrieval Receptor for Endoplasmic Reticulum Membrane Proteins, Is Dynamically Localized to the Golgi Apparatus by Coatomer. J. Cell Biol. 2001, 152, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Flanagan, J.J.; Barlowe, C. Sec22p export from the endoplasmic reticulum is independent of SNARE pairing. J. Biol. Chem. 2004, 279, 27225–27232. [Google Scholar] [CrossRef]
- Taubenschmid, J.; Stadlmann, J.; Jost, M.; Klokk, T.I.; Rillahan, C.D.; Leibbrandt, A.; Mechtler, K.; Paulson, J.C.; Jude, J.; Zuber, J.; et al. A vital sugar code for ricin toxicity. Cell Res. 2017, 27, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.C.; Roberts, L.M.; Römisch, K.; Davey, J.; Wolf, D.H.; Lord, J.M. Ricin A chain utilises the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett. 1999, 459, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Parikh, B.A.; Tortora, A.; Li, X.-P.; Tumer, N.E. Ricin inhibits activation of the unfolded protein response by preventing splicing of the HAC1 mRNA. J. Biol. Chem. 2008, 283, 6145–6153. [Google Scholar] [CrossRef]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.H.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J. 2004, 383, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Redmann, V.; Oresic, K.; Tortorella, L.L.; Cook, J.P.; Lord, M.; Tortorella, D. Dislocation of Ricin Toxin A Chains in Human Cells Utilizes Selective Cellular Factors. J. Biol. Chem. 2011, 286, 21231–21238. [Google Scholar] [CrossRef] [Green Version]
- Di Cola, A.; Frigerio, L.; Lord, J.M.; Ceriotti, A.; Roberts, L.M. Ricin A chain without its partner B chain is degraded after retrotranslocation from the endoplasmic reticulum to the cytosol in plant cells. Proc. Natl. Acad. Sci. USA 2001, 98, 14726–14731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemmill, T.R.; Trimble, R.B. Overview of N- and O-linked oligosaccharide structures found in various yeast species. Biochim. Biophys. Acta Gen. Subj. 1999, 1426, 227–237. [Google Scholar] [CrossRef]
- Lamb, F.I.; Roberts, L.M.; Lord, J.M. Nucleotide sequence of cloned cDNA coding for preproricin. Eur. J. Biochem. 1985, 148, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Rapak, A.; Olsnes, S. Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 1999, 274, 34443–34449. [Google Scholar] [CrossRef]
- Spooner, R.A.; Lord, J.M. How ricin and Shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 2012, 357, 19–40. [Google Scholar]
- Nowakowska-Gołacka, J.; Sominka, H.; Sowa-Rogozińska, N.; Słomińska-Wojewódzka, M. Toxins Utilize the Endoplasmic Reticulum-Associated Protein Degradation Pathway in Their Intoxication Process. Int. J. Mol. Sci. 2019, 20, 1307. [Google Scholar] [CrossRef]
- Mohanraj, D.; Ramakrishnan, S. Cytotoxic effects of ricin without an interchain disulfide bond: Genetic modification and chemical crosslinking studies. Biochim. Biophys. Acta Gen. Subj. 1995, 1243, 399–406. [Google Scholar] [CrossRef]
- Freedman, R.B.; Hirst, T.R.; Tuite, M.F. Protein disulphide isomerase: Building bridges in protein folding. Trends Biochem. Sci. 1994, 19, 331–336. [Google Scholar] [CrossRef]
- Bulleid, N.J. Disulfide Bond Formation in the Mammalian Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2012, 4, a013219. [Google Scholar] [CrossRef]
- Bellisola, G.; Fracasso, G.; Ippoliti, R.; Menestrina, G.; Rosén, A.; Soldà, S.; Udali, S.; Tomazzolli, R.; Tridente, G.; Colombatti, M. Reductive activation of ricin and ricin A-chain immunotoxins by protein disulfide isomerase and thioredoxin reductase. Biochem. Pharmacol. 2004, 67, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.T.; Westby, M.; Roberts, L.M.; Gould, J.H.; Colman, A.; Lord, J.M. Recombinant proricin binds galactose but does not depurinate 28 S ribosomal RNA. FEBS Lett. 1989, 255, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Pasetto, M.; Barison, E.; Castagna, M.; Della Cristina, P.; Anselmi, C.; Colombatti, M. Reductive activation of type 2 ribosome-inactivating proteins is promoted by transmembrane thioredoxin-related protein. J. Biol. Chem. 2012, 287, 7367–7373. [Google Scholar] [CrossRef]
- Argent, R.H.; Roberts, L.M.; Wales, R.; Robertus, J.D.; Lord, J.M. Introduction of a disulfide bond into ricin A chain decreases the cytotoxicity of the ricin holotoxin. J. Biol. Chem. 1994, 269, 26705–26710. [Google Scholar]
- Argent, R.H.; Parrott, A.M.; Day, P.J.; Roberts, L.M.; Stockley, P.G.; Lord, J.M.; Radford, S.E. Ribosome-mediated folding of partially unfolded ricin A-chain. J. Biol. Chem. 2000, 275, 9263–9269. [Google Scholar] [CrossRef]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Braakman, I.; Hebert, D.N. Protein Folding in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef]
- Karagöz, G.E.; Acosta-Alvear, D.; Walter, P. The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2019, a033886. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Ye, Y.; Rapoport, T.A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 2002, 3, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Gregers, T.F.; Skånland, S.S.; Wälchli, S.; Bakke, O.; Sandvig, K. BiP negatively affects ricin transport. Toxins 2013, 5, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kotler, J.L.M.; Liu, S.; Street, T.O. The ER chaperones BiP and Grp94 selectively associate when BiP is in the ADP conformation. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Hart, P.J.; Cook, J.P.; Pietroni, P.; Rogon, C.; Höhfeld, J.; Roberts, L.M.; Lord, J.M. Cytosolic chaperones influence the fate of a toxin dislocated from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 17408–17413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearse, B.R.; Hebert, D.N. Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 684–693. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, M.; Srinivas, H.; Kandiah, E.; Gemma, E.; Ellgaard, L.; Oscarson, S.; Helenius, A.; Surolia, A. Interactions of Substrate with Calreticulin, an Endoplasmic Reticulum Chaperone. J. Biol. Chem. 2003, 278, 6194–6200. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Wada, I.; Hasegawa, K.; Yorihuzi, T.; Tremblay, L.O.; Herscovics, A.; Nagata, K. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001, 2, 415–422. [Google Scholar] [CrossRef]
- Mast, S.W.; Diekman, K.; Karaveg, K.; Davis, A.; Sifers, R.N.; Moremen, K.W. Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology 2005, 15, 421–436. [Google Scholar] [CrossRef]
- Hirao, K.; Natsuka, Y.; Tamura, T.; Wada, I.; Morito, D.; Natsuka, S.; Romero, P.; Sleno, B.; Tremblay, L.O.; Herscovics, A.; et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006, 281, 9650–9658. [Google Scholar] [CrossRef]
- Olivari, S.; Molinari, M. Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS Lett. 2007, 581, 3658–3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominska-Wojewodzka, M.; Gregers, T.F.; Wälchli, S.; Sandvig, K. EDEM Is Involved in Retrotranslocation of Ricin from the Endoplasmic Reticulum to the Cytosol. Mol. Biol. Cell 2006, 17, 1664–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Słomińska-Wojewódzka, M.; Pawlik, A.; Sokołowska, I.; Antoniewicz, J.; Węgrzyn, G.; Sandvig, K. The role of EDEM2 compared with EDEM1 in ricin transport from the endoplasmic reticulum to the cytosol. Biochem. J. 2014, 457, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Słomińska-Wojewódzka, M.; Sandvig, K. The Role of Lectin-Carbohydrate Interactions in the Regulation of ER-Associated Protein Degradation. Molecules 2015, 20, 9816–9846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sominka, H.; Nowakowska-Gołacka, J.; Sowa-Rogozińska, N.; Słomińska-Wojewódzka, M. The role of EDEM3 in ricin cytotoxicity and its transport from the ER to the cytosol. Unpublished. Manuscript in preparation.
- Mayerhofer, P.U.; Cook, J.P.; Wahlman, J.; Pinheiro, T.T.J.; Moore, K.A.H.; Lord, J.M.; Johnson, A.E.; Roberts, L.M. Ricin A Chain Insertion into Endoplasmic Reticulum Membranes Is Triggered by a Temperature Increase to 37 °C. J. Biol. Chem. 2009, 284, 10232–10242. [Google Scholar] [CrossRef] [PubMed]
- Katzin, B.J.; Collins, E.J.; Robertus, J.D. Structure of ricin A-chain at 2.5 A. Proteins 1991, 10, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Li, X.-P.; Tumer, N.E. N-glycosylation does not affect the catalytic activity of ricin a chain but stimulates cytotoxicity by promoting its transport out of the endoplasmic reticulum. Traffic 2012, 13, 1508–1521. [Google Scholar] [CrossRef] [PubMed]
- Leto, D.E.; Morgens, D.W.; Zhang, L.; Walczak, C.P.; Elias, J.E.; Bassik, M.C.; Kopito, R.R. Genome-wide CRISPR Analysis Identifies Substrate-Specific Conjugation Modules in ER-Associated Degradation. Mol. Cell 2019, 73, 377–389. [Google Scholar] [CrossRef]
- Pilon, M.; Schekman, R.; Römisch, K. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 1997, 16, 4540–4548. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.; Wolf, D.H. Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 2009, 28, 2874–2884. [Google Scholar] [CrossRef] [Green Version]
- Tretter, T.; Pereira, F.P.; Ulucan, O.; Helms, V.; Allan, S.; Kalies, K.-U.; Römisch, K. ERAD and protein import defects in a sec61 mutant lacking ER-lumenal loop 7. BMC Cell Biol. 2013, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.-L.; Römisch, K. Proteasome 19S RP Binding to the Sec61 Channel Plays a Key Role in ERAD. PLoS ONE 2015, 10, e0117260. [Google Scholar] [CrossRef] [PubMed]
- Römisch, K. A Case for Sec61 Channel Involvement in ERAD. Trends Biochem. Sci. 2017, 42, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Ploegh, H.L. A membrane protein required for dislocation of misfolded proteins from the ER. Nature 2004, 429, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Ploegh, H.L. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14296–14301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, Y.; Okada, T.; Yoshida, H.; Kaufman, R.J.; Nagata, K.; Mori, K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J. Cell Biol. 2006, 172, 383–393. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Ge, Y.; Zhang, J.; Cao, Y.; Xing, J.; Su, D.; Huang, Y.; Li, M.; Qu, S.; Sun, F.; et al. Derlin-1 promotes ubiquitylation and degradation of the epithelial Na+ channel, ENaC. J. Cell Sci. 2017, 130, 1027–1036. [Google Scholar]
- Mehnert, M.; Sommer, T.; Jarosch, E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol. 2014, 16, 77–86. [Google Scholar] [CrossRef]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar] [CrossRef]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 2010, 143, 579–591. [Google Scholar] [CrossRef]
- Stein, A.; Ruggiano, A.; Carvalho, P.; Rapoport, T.A. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 2014, 158, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, R.D.; Rapoport, T.A. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 2016, 166, 394–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoebel, S.; Mi, W.; Stein, A.; Ovchinnikov, S.; Pavlovicz, R.; DiMaio, F.; Baker, D.; Chambers, M.G.; Su, H.; Li, D.; et al. Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 2017, 548, 352–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Sowa-Rogozińska, N.; Słomińska-Wojewódzka, M. The role of Sec61 in ricin transport from the ER to the cytosol. Unpublished. Manuscript in preparation.
- Sowa-Rogozińska, N.; Sominka, H.; Słomińska-Wojewódzka, M. The role of Derlin proteins in ricin transport from the ER to the cytosol. Unpublished. Manuscript in preparation.
- Dang, H.; Klokk, T.I.; Schaheen, B.; McLaughlin, B.M.; Thomas, A.J.; Durns, T.A.; Bitler, B.G.; Sandvig, K.; Fares, H. Derlin-dependent retrograde transport from endosomes to the Golgi apparatus. Traffic 2011, 12, 1417–1431. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Spooner, R.A.; Allen, S.C.H.; Guise, C.P.; Ladds, G.; Schnöder, T.; Schmitt, M.J.; Lord, J.M.; Roberts, L.M. Folding-competent and folding-defective forms of ricin A chain have different fates after retrotranslocation from the endoplasmic reticulum. Mol. Biol. Cell 2010, 21, 2543–2554. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, A.; Dixon, S.D.; Tamilselvam, B.; Kim, E.J.-K.; Gargi, A.; Kulik, J.C.; Damoiseaux, R.; Blanke, S.R.; Bradley, K.A. Cytolethal distending toxins require components of the ER-associated degradation pathway for host cell entry. PLoS Pathog. 2014, 10, e1004295. [Google Scholar] [CrossRef]
- Pietroni, P.; Vasisht, N.; Cook, J.P.; Roberts, D.M.; Lord, J.M.; Hartmann-Petersen, R.; Roberts, L.M.; Spooner, R.A. The proteasome cap RPT5/Rpt5p subunit prevents aggregation of unfolded ricin A chain. Biochem. J. 2013, 453, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Di Cola, A.; Frigerio, L.; Lord, J.M.; Roberts, L.M.; Ceriotti, A. Endoplasmic Reticulum-Associated Degradation of Ricin A Chain Has Unique and Plant-Specific Features. Plant Physiol. 2005, 137, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Deeks, E.D.; Cook, J.P.; Day, P.J.; Smith, D.C.; Roberts, L.M.; Lord, J.M. The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry 2002, 41, 3405–3413. [Google Scholar] [CrossRef] [PubMed]
- Abujarour, R.J.; Dalal, S.; Hanson, P.I.; Draper, R.K. p97 Is in a complex with cholera toxin and influences the transport of cholera toxin and related toxins to the cytoplasm. J. Biol. Chem. 2005, 280, 15865–15871. [Google Scholar] [CrossRef] [PubMed]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodighiero, C.; Tsai, B.; Rapoport, T.A.; Lencer, W.I. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002, 3, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Lipson, C.; Alalouf, G.; Bajorek, M.; Rabinovich, E.; Atir-Lande, A.; Glickman, M.; Bar-Nun, S. A proteasomal ATPase contributes to dislocation of endoplasmic reticulum-associated degradation (ERAD) substrates. J. Biol. Chem. 2008, 283, 7166–7175. [Google Scholar] [CrossRef] [PubMed]
- Odunuga, O.O.; Longshaw, V.M.; Blatch, G.L. Hop: More than an Hsp70/Hsp90 adaptor protein. Bioessays 2004, 26, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-P.; Grela, P.; Krokowski, D.; Tchórzewski, M.; Tumer, N.E. Pentameric organization of the ribosomal stalk accelerates recruitment of ricin a chain to the ribosome for depurination. J. Biol. Chem. 2010, 285, 41463–41471. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.L.; Sloma, M.S.; Nygård, O. Conformational changes in the structure of domains II and V of 28S rRNA in ribosomes treated with the translational inhibitors ricin or alpha-sarcin. Biochim. Biophys. Acta 2002, 1577, 53–62. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar]
- May, K.L.; Yan, Q.; Tumer, N.E. Targeting ricin to the ribosome. Toxicon 2013, 69, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, J.-C.; Li, X.-P.; Remacha, M.; Ballesta, J.P.G.; Tumer, N.E. The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae. Mol. Microbiol. 2008, 70, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- May, K.L.; Li, X.-P.; Martínez-Azorín, F.; Ballesta, J.P.G.; Grela, P.; Tchórzewski, M.; Tumer, N.E. The P1/P2 proteins of the human ribosomal stalk are required for ribosome binding and depurination by ricin in human cells. FEBS J. 2012, 279, 3925–3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grela, P.; Sawa-Makarska, J.; Gordiyenko, Y.; Robinson, C.V.; Grankowski, N.; Tchórzewski, M. Structural properties of the human acidic ribosomal P proteins forming the P1-P2 heterocomplex. J. Biochem. 2008, 143, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-M.; Yu, C.W.-H.; Chiu, T.Y.-H.; Sze, K.-H.; Shaw, P.-C.; Wong, K.-B. Solution structure of the dimerization domain of the eukaryotic stalk P1/P2 complex reveals the structural organization of eukaryotic stalk complex. Nucleic Acids Res. 2012, 40, 3172–3182. [Google Scholar] [CrossRef] [PubMed]
- Grela, P.; Li, X.-P.; Horbowicz, P.; Dźwierzyńska, M.; Tchórzewski, M.; Tumer, N.E. Human ribosomal P1-P2 heterodimer represents an optimal docking site for ricin A chain with a prominent role for P1 C-terminus. Sci. Rep. 2017, 7, 5608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grela, P.; Li, X.-P.; Tchórzewski, M.; Tumer, N.E. Functional divergence between the two P1-P2 stalk dimers on the ribosome in their interaction with ricin A chain. Biochem. J. 2014, 460, 59–67. [Google Scholar] [CrossRef]
- Grela, P.; Bernadó, P.; Svergun, D.; Kwiatowski, J.; Abramczyk, D.; Grankowski, N.; Tchórzewski, M. Structural relationships among the ribosomal stalk proteins from the three domains of life. J. Mol. Evol. 2008, 67, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-P.; Chiou, J.-C.; Remacha, M.; Ballesta, J.P.G.; Tumer, N.E. A two-step binding model proposed for the electrostatic interactions of ricin A chain with ribosomes. Biochemistry 2009, 48, 3853–3863. [Google Scholar] [CrossRef] [PubMed]
- Tumer, N.E.; Li, X.-P. Interaction of ricin and Shiga toxins with ribosomes. Curr. Top. Microbiol. Immunol. 2012, 357, 1–18. [Google Scholar]
- Fan, X.; Zhu, Y.; Wang, C.; Niu, L.; Teng, M.; Li, X. Structural insights into the interaction of the ribosomal P stalk protein P2 with a type II ribosome-inactivating protein ricin. Sci. Rep. 2016, 6, 37803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.-W.; Tang, Y.-S.; Sze, S.-Y.; Zhu, Z.-N.; Wong, K.-B.; Shaw, P.-C. Crystal Structure of Ribosome-Inactivating Protein Ricin A Chain in Complex with the C-Terminal Peptide of the Ribosomal Stalk Protein P2. Toxins 2016, 8, 296. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Dansako, H.; Asada, N.; Sakai, M.; Funatsu, G. Effects of chemical modification of arginine residues outside the active site cleft of ricin A-chain on its RNA N-glycosidase activity for ribosomes. Biosci. Biotechnol. Biochem. 1994, 58, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.J.; Fülöp, V.; Day, P.J.; Lord, J.M. The effect of mutations surrounding and within the active site on the catalytic activity of ricin A chain. Eur. J. Biochem. 2004, 271, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, X.-P.; Chen, B.Y.; Tumer, N.E. Ricin uses arginine 235 as an anchor residue to bind to P-proteins of the ribosomal stalk. Sci. Rep. 2017, 7, 42912. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-P.; Kahn, P.C.; Kahn, J.N.; Grela, P.; Tumer, N.E. Arginine residues on the opposite side of the active site stimulate the catalysis of ribosome depurination by ricin A chain by interacting with the P-protein stalk. J. Biol. Chem. 2013, 288, 30270–30284. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-P.; Tumer, N.E. Differences in Ribosome Binding and Sarcin/Ricin Loop Depurination by Shiga and Ricin Holotoxins. Toxins 2017, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Jetzt, A.E.; Li, X.-P.; Tumer, N.E.; Cohick, W.S. Toxicity of ricin A chain is reduced in mammalian cells by inhibiting its interaction with the ribosome. Toxicol. Appl. Pharmacol. 2016, 310, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Hedblom, M.L.; Cawley, D.B.; Boguslawski, S.; Houston, L.L. Binding of ricin A chain to rat liver ribosomes: Relationship to ribosome inactivation. J. Supramol. Struct. 1978, 9, 253–268. [Google Scholar] [CrossRef]
- Honjo, E.; Watanabe, K.; Tsukamoto, T. Real-time kinetic analyses of the interaction of ricin toxin A-chain with ribosomes prove a conformational change involved in complex formation. J. Biochem. 2002, 131, 267–275. [Google Scholar] [CrossRef]
- Dai, J.; Zhao, L.; Yang, H.; Guo, H.; Fan, K.; Wang, H.; Qian, W.; Zhang, D.; Li, B.; Wang, H.; et al. Identification of a novel functional domain of ricin responsible for its potent toxicity. J. Biol. Chem. 2011, 286, 12166–12171. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.N.; Wool, I.G. Determination by systematic deletion of the amino acids essential for catalysis by ricin A chain. Proc. Natl. Acad. Sci. USA 1992, 89, 4869–4873. [Google Scholar] [CrossRef] [PubMed]
- Day, P.J.; Ernst, S.R.; Frankel, A.E.; Monzingo, A.F.; Pascal, J.M.; Molina-Svinth, M.C.; Robertus, J.D. Structure and activity of an active site substitution of ricin A chain. Biochemistry 1996, 35, 11098–11103. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-P.; Kahn, J.N.; Tumer, N.E. Peptide Mimics of the Ribosomal P Stalk Inhibit the Activity of Ricin A Chain by Preventing Ribosome Binding. Toxins 2018, 10, 371. [Google Scholar] [CrossRef]
- Lewis, J.L.; Shields, K.A.; Chong, D.C. Detection and quantification of ricin-mediated 28S ribosomal depurination by digital droplet PCR. Anal. Biochem. 2018, 563, 15–19. [Google Scholar] [CrossRef]
- Falach, R.; Sapoznikov, A.; Gal, Y.; Israeli, O.; Leitner, M.; Seliger, N.; Ehrlich, S.; Kronman, C.; Sabo, T. Quantitative profiling of the in vivo enzymatic activity of ricin reveals disparate depurination of different pulmonary cell types. Toxicol. Lett. 2016, 258, 11–19. [Google Scholar] [CrossRef]
- Meneguelli de Souza, L.C.; de Carvalho, L.P.; Araújo, J.S.; de Melo, E.J.T.; Machado, O.L.T. Cell toxicity by ricin and elucidation of mechanism of Ricin inactivation. Int. J. Biol. Macromol. 2018, 113, 821–828. [Google Scholar] [CrossRef]
- Sandvig, K.; van Deurs, B. Toxin-induced cell lysis: Protection by 3-methyladenine and cycloheximide. Exp. Cell Res. 1992, 200, 253–262. [Google Scholar] [CrossRef]
- Bagaria, S.; Karande, A. Abrin and Immunoneutralization: A Review. Toxinology 2014. [Google Scholar] [CrossRef]
- Bingen, A.; Creppy, E.E.; Gut, J.P.; Dirheimer, G.; Kirn, A. The Kupffer cell is the first target in ricin-induced hepatitis. J. Submicrosc. Cytol. 1987, 19, 247–256. [Google Scholar]
- Leek, M.D.; Griffiths, G.D.; Green, M.A. Intestinal pathology following intramuscular ricin poisoning. J. Pathol. 1989, 159, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, C.L.; Pitt, M.L. Lesions of acute inhaled lethal ricin intoxication in rhesus monkeys. Vet. Pathol. 1996, 33, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Soler-Rodríguez, A.M.; Ghetie, M.A.; Oppenheimer-Marks, N.; Uhr, J.W.; Vitetta, E.S. Ricin A-chain and ricin A-chain immunotoxins rapidly damage human endothelial cells: Implications for vascular leak syndrome. Exp. Cell Res. 1993, 206, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Kochi, S.K.; Collier, R.J. DNA fragmentation and cytolysis in U937 cells treated with diphtheria toxin or other inhibitors of protein synthesis. Exp. Cell Res. 1993, 208, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Peumans, W.J.; Hao, Q.; Van Damme, E.J. Ribosome-inactivating proteins from plants: More than RNA N-glycosidases? FASEB J. 2001, 15, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Brigotti, M.; Alfieri, R.; Sestili, P.; Bonelli, M.; Petronini, P.G.; Guidarelli, A.; Barbieri, L.; Stirpe, F.; Sperti, S. Damage to nuclear DNA induced by Shiga toxin 1 and ricin in human endothelial cells. FASEB J. 2002, 16, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Pestka, J.J. Comparative induction of 28S ribosomal RNA cleavage by ricin and the trichothecenes deoxynivalenol and T-2 toxin in the macrophage. Toxicol. Sci. 2008, 105, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.S.; Bae, H.K.; Li, J.C.B.; Lau, A.S.; Pestka, J.J. Double-stranded RNA-activated protein kinase mediates induction of interleukin-8 expression by deoxynivalenol, Shiga toxin 1, and ricin in monocytes. Toxicol. Sci. 2008, 105, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. The induction of apoptosis by Shiga toxins and ricin. Curr. Top. Microbiol. Immunol. 2012, 357, 137–178. [Google Scholar] [PubMed]
- Komatsu, N.; Oda, T.; Muramatsu, T. Involvement of both caspase-like proteases and serine proteases in apoptotic cell death induced by ricin, modeccin, diphtheria toxin, and pseudomonas toxin. J. Biochem. 1998, 124, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Lea, N.; Lord, J.M.; Roberts, L.M.; Milford, D.V.; Taylor, C.M. Comparison of ribosome-inactivating proteins in the induction of apoptosis. Toxicol. Lett. 1997, 91, 121–127. [Google Scholar] [CrossRef]
- Rao, P.V.L.; Jayaraj, R.; Bhaskar, A.S.B.; Kumar, O.; Bhattacharya, R.; Saxena, P.; Dash, P.K.; Vijayaraghavan, R. Mechanism of ricin-induced apoptosis in human cervical cancer cells. Biochem. Pharmacol. 2005, 69, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Jetzt, A.E.; Cheng, J.-S.; Tumer, N.E.; Cohick, W.S. Ricin A-chain requires c-Jun N-terminal kinase to induce apoptosis in nontransformed epithelial cells. Int. J. Biochem. Cell Biol. 2009, 41, 2503–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, N.; Nakagawa, M.; Oda, T.; Muramatsu, T. Depletion of intracellular NAD(+) and ATP levels during ricin-induced apoptosis through the specific ribosomal inactivation results in the cytolysis of U937 cells. J. Biochem. 2000, 128, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-H.; Shih, S.-F.; Lin, J.-Y. Ricin Triggers Apoptotic Morphological Changes through Caspase-3 Cleavage of BAT3. J. Biol. Chem. 2004, 279, 19264–19275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, O.; Yew, D.T.W.; Ng, T.B.; Yuan, L.; Kwong, W.H. Different in vitro toxicities of structurally similar type I ribosome-inactivating proteins (RIPs). Toxicol. In Vitro 2010, 24, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Mansfield, E.; Pastan, I. Effects of BCL-2 overexpression on the sensitivity of MCF-7 breast cancer cells to ricin, diphtheria and Pseudomonas toxin and immunotoxins. Apoptosis 1997, 2, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Zhai, Q.; Liu, W.; Liu, X. An insight into the mechanism of cytotoxicity of ricin to hepatoma cell: Roles of Bcl-2 family proteins, caspases, Ca(2+)-dependent proteases and protein kinase C. J. Cell. Biochem. 2001, 81, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Oda, T.; Muramatsu, T. Resistance against ricin-induced apoptosis in a brefeldin A-resistant mutant cell line (BER-40) of Vero cells. J. Biochem. 2002, 132, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: Activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell Biol. 1997, 17, 3373–3381. [Google Scholar] [CrossRef]
- Korcheva, V.; Wong, J.; Corless, C.; Iordanov, M.; Magun, B. Administration of ricin induces a severe inflammatory response via nonredundant stimulation of ERK, JNK, and P38 MAPK and provides a mouse model of hemolytic uremic syndrome. Am. J. Pathol. 2005, 166, 323–339. [Google Scholar] [CrossRef]
- Gonzalez, T.V.; Farrant, S.A.; Mantis, N.J. Ricin induces IL-8 secretion from human monocyte/macrophages by activating the p38 MAP kinase pathway. Mol. Immunol. 2006, 43, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Korcheva, V.; Wong, J.; Lindauer, M.; Jacoby, D.B.; Iordanov, M.S.; Magun, B. Role of Apoptotic Signaling Pathways in Regulation of Inflammatory Responses to Ricin in Primary Murine Macrophages. Mol. Immunol. 2007, 44, 2761–2771. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Korcheva, V.; Jacoby, D.B.; Magun, B.E. Proinflammatory responses of human airway cells to ricin involve stress-activated protein kinases and NF-kappaB. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1385–L1394. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, C.; Nishikawa, K.; Zeng, X.-T.; Katayama, Y.; Natori, Y.; Komatsu, N.; Oda, T.; Natori, Y. Induction of cytokines by toxins that have an identical RNA N-glycosidase activity: Shiga toxin, ricin, and modeccin. Biochim. Biophys. Acta 2004, 1671, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, M.L.; Wong, J.; Iwakura, Y.; Magun, B.E. Pulmonary inflammation triggered by ricin toxin requires macrophages and IL-1 signaling. J. Immunol. 2009, 183, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, M.; Wong, J.; Magun, B. Ricin Toxin Activates the NALP3 Inflammasome. Toxins 2010, 2, 1500–1514. [Google Scholar] [CrossRef] [Green Version]
- Gal, Y.; Mazor, O.; Falach, R.; Sapoznikov, A.; Kronman, C.; Sabo, T. Treatments for Pulmonary Ricin Intoxication: Current Aspects and Future Prospects. Toxins 2017, 9, 311. [Google Scholar] [CrossRef]
- Jandhyala, D.M.; Ahluwalia, A.; Obrig, T.; Thorpe, C.M. ZAK: A MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell. Microbiol. 2008, 10, 1468–1477. [Google Scholar] [CrossRef]
- Higuchi, S.; Tamura, T.; Oda, T. Cross-talk between the pathways leading to the induction of apoptosis and the secretion of tumor necrosis factor-alpha in ricin-treated RAW 264.7 cells. J. Biochem. 2003, 134, 927–933. [Google Scholar] [CrossRef]
- Wang, C.-T.; Jetzt, A.E.; Cheng, J.-S.; Cohick, W.S. Inhibition of the Unfolded Protein Response by Ricin A-Chain Enhances Its Cytotoxicity in Mammalian Cells. Toxins 2011, 3, 453–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horrix, C.; Raviv, Z.; Flescher, E.; Voss, C.; Berger, M.R. Plant ribosome-inactivating proteins type II induce the unfolded protein response in human cancer cells. Cell. Mol. Life Sci. 2011, 68, 1269–1281. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, L.; Brigotti, M.; Perocco, P.; Carnicelli, D.; Ciani, M.; Mercatali, L.; Stirpe, F. Ribosome-inactivating proteins depurinate poly(ADP-ribosyl) ated poly(ADP-ribose) polymerase and have transforming activity for 3T3 fibroblasts. FEBS Lett. 2003, 538, 178–182. [Google Scholar] [CrossRef]
- Oda, T.; Iwaoka, J.; Komatsu, N.; Muramatsu, T. Involvement of N-acetylcysteine-sensitive pathways in ricin-induced apoptotic cell death in U937 cells. Biosci. Biotechnol. Biochem. 1999, 63, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Komatsu, N.; Muramatsu, T. Inhibitory effect of dideoxyforskolin on cell death induced by ricin, modeccin, diphtheria toxin, and Pseudomonas toxin in MDCK cells. Cell Struct. Funct. 1997, 22, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Sadakata, N.; Oda, T.; Muramatsu, T. Role of zinc ions in ricin-induced apoptosis in U937 cells. Toxicol. Lett. 2002, 132, 141–151. [Google Scholar] [CrossRef]
- Authier, F.; Djavaheri-Mergny, M.; Lorin, S.; Frénoy, J.-P.; Desbuquois, B. Fate and action of ricin in rat liver in vivo: Translocation of endocytosed ricin into cytosol and induction of intrinsic apoptosis by ricin B-chain. Cell. Microbiol. 2016, 18, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- Kitazumi, I.; Tsukahara, M. Regulation of DNA fragmentation: The role of caspases and phosphorylation. FEBS J. 2011, 278, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Desmots, F.; Russell, H.R.; Michel, D.; McKinnon, P.J. Scythe regulates apoptosis-inducing factor stability during endoplasmic reticulum stress-induced apoptosis. J. Biol. Chem. 2008, 283, 3264–3271. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Lin, A.; Minden, A.; Martinetto, H.; Claret, F.X.; Lange-Carter, C.; Mercurio, F.; Johnson, G.L.; Karin, M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science 1995, 268, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ma, Y.; Pagliari, L.J.; Perlman, H.; Yu, C.; Lin, A.; Pope, R.M. TNF-alpha-induced apoptosis of macrophages following inhibition of NF-kappa B: A central role for disruption of mitochondria. J. Immunol. 2004, 172, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Bubici, C.; Zazzeroni, F.; Pham, C.G.; Kuntzen, C.; Knabb, J.R.; Dean, K.; Franzoso, G. The NF-kappaB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease. Cell Death Differ. 2006, 13, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; Papa, F.R.; Walter, P. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2006, 22, 487–508. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Guerra-Moreno, A.; Ang, J.; Welsch, H.; Jochem, M.; Hanna, J. Regulation of the unfolded protein response in yeast by oxidative stress. FEBS Lett. 2019, 593, 1080–1088. [Google Scholar] [CrossRef]
- Barbieri, L.; Valbonesi, P.; Bonora, E.; Gorini, P.; Bolognesi, A.; Stirpe, F. Polynucleotide: Adenosine glycosidase activity of ribosome-inactivating proteins: Effect on DNA, RNA and poly(A). Nucleic Acids Res. 1997, 25, 518–522. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl) ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef]
- Sestili, P.; Alfieri, R.; Carnicelli, D.; Martinelli, C.; Barbieri, L.; Stirpe, F.; Bonelli, M.; Petronini, P.G.; Brigotti, M. Shiga toxin 1 and ricin inhibit the repair of H2O2-induced DNA single strand breaks in cultured mammalian cells. DNA Repair 2005, 4, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Gong, Y.; Ma, H.; An, C.; Chen, D.; Chen, Z.L. Reactive oxygen species involved in trichosanthin-induced apoptosis of human choriocarcinoma cells. Biochem. J. 2001, 355, 653–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsden, C.S.; Drayson, M.T.; Bell, E.B. The toxicity, distribution and excretion of ricin holotoxin in rats. Toxicology 1989, 55, 161–171. [Google Scholar] [CrossRef]
- Xu, N.; Yuan, H.; Liu, W.; Li, S.; Liu, Y.; Wan, J.; Li, X.; Zhang, R.; Chang, Y. Activation of RAW264.7 mouse macrophage cells in vitro through treatment with recombinant ricin toxin-binding subunit B: Involvement of protein tyrosine, NF-κB and JAK-STAT kinase signaling pathways. Int. J. Mol. Med. 2013, 32, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, A.M. The collected papers of Paul Ehrlich: Why was volume 4 never published? Bull. Hist. Med. 2002, 76, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Vitetta, E.S.; Thorpe, P.E.; Uhr, J.W. Immunotoxins: Magic bullets or misguided missiles? Immunol. Today 1993, 14, 252–259. [Google Scholar] [CrossRef]
- Brinkmann, U.; Pastan, I. Immunotoxins against cancer. Biochim. Biophys. Acta 1994, 1198, 27–45. [Google Scholar] [CrossRef]
- Weidle, U.H.; Tiefenthaler, G.; Schiller, C.; Weiss, E.H.; Georges, G.; Brinkmann, U. Prospects of bacterial and plant protein-based immunotoxins for treatment of cancer. Cancer Genomics Proteomics 2014, 11, 25–38. [Google Scholar] [PubMed]
- Munir, I.; Naseer, R.; Saleem, M.; Mahmood, A.; Sultana, A. Immunotoxins, an Advance tool for Cancer Treatment: Review and update. Acta Pol. Pharm. Drug Res. 2018, 75, 1267–1277. [Google Scholar] [CrossRef]
- Polito, L.; Djemil, A.; Bortolotti, M. Plant Toxin-Based Immunotoxins for Cancer Therapy: A Short Overview. Biomedicines 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Goldmacher, V.S.; Collinson, A.R.; Nadler, L.M.; Blättler, W.A. An immunotoxin prepared with blocked ricin: A natural plant toxin adapted for therapeutic use. Cancer Res. 1991, 51, 6236–6242. [Google Scholar]
- Krolick, K.A.; Villemez, C.; Isakson, P.; Uhr, J.W.; Vitetta, E.S. Selective killing of normal or neoplastic B cells by antibodies coupled to the A chain of ricin. Proc. Natl. Acad. Sci. USA 1980, 77, 5419–5423. [Google Scholar] [CrossRef] [PubMed]
- Bourrie, B.J.; Casellas, P.; Blythman, H.E.; Jansen, F.K. Study of the plasma clearance of antibody—Ricin-A-chain immunotoxins. Evidence for specific recognition sites on the A chain that mediate rapid clearance of the immunotoxin. Eur. J. Biochem. 1986, 155, 1–10. [Google Scholar] [CrossRef]
- Fulton, R.J.; Uhr, J.W.; Vitetta, E. In Vivo Therapy of the BCL1 Tumor: Effect of Immunotoxin Valency and Deglycosylation of the Ricin A Chain. Cancer Res. 1988, 48, 2626–2631. [Google Scholar]
- Blakey, D.C.; Watson, G.; Knowles, P.P.; Thorpe, P. Effect of chemical deglycosylation of ricin A chain on the in vivo fate and cytotoxic activity of an immunotoxin composed of ricin A chain and anti-Thy 1.1 antibody. Cancer Res. 1987, 47, 947–952. [Google Scholar]
- Street, N.E.; Fulton, R.J.; Sanders, V.M.; Vitetta, E.S. Inhibition of the helper function of murine T cells with Fab’-anti-L3T4 ricin A chain immunotoxin. J. Immunol. 1987, 139, 1734–1738. [Google Scholar]
- Li, C.; Yan, R.; Yang, Z.; Wang, H.; Zhang, R.; Chen, H.; Wang, J. BCMab1-Ra, a novel immunotoxin that BCMab1 antibody coupled to Ricin A chain, can eliminate bladder tumor. Oncotarget 2017, 8, 46704–46705. [Google Scholar] [CrossRef]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Parveen, S.; Misra, R.; Sahoo, S.K. Nanoparticles: A boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine 2012, 8, 147–166. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.W.; Bae, Y.H. Odyssey of a cancer nanoparticle: From injection site to site of action. Nano Today 2012, 7, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Skotland, T.; Iversen, T.-G.; Sandvig, K. Development of nanoparticles for clinical use. Nanomedicine 2014, 9, 1295–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekle, C.; van Deurs, B.; Sandvig, K.; Iversen, T.-G. Cellular trafficking of quantum dot-ligand bioconjugates and their induction of changes in normal routing of unconjugated ligands. Nano Lett. 2008, 8, 1858–1865. [Google Scholar] [CrossRef]
- Iversen, T.-G.; Frerker, N.; Sandvig, K. Quantum dot bioconjugates: Uptake into cells and induction of changes in normal cellular transport. In Colloidal Quantum Dots for Biomedical Applications IV; International Society for Optics and Photonics: Bellingham, WA, USA, 2009; Volume 7189, p. 71890T. [Google Scholar]
- Li, Y.; Liu, W.; Sun, C.; Zheng, M.; Zhang, J.; Liu, B.; Wang, Y.; Xie, Z.; Xu, N. Hybrids of carbon dots with subunit B of ricin toxin for enhanced immunomodulatory activity. J. Colloid Interface Sci. 2018, 523, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Díaz, R.; Pallarès, V.; Cano-Garrido, O.; Serna, N.; Sánchez-García, L.; Falgàs, A.; Pesarrodona, M.; Unzueta, U.; Sánchez-Chardi, A.; Sánchez, J.M.; et al. Selective CXCR4+ Cancer Cell Targeting and Potent Antineoplastic Effect by a Nanostructured Version of Recombinant Ricin. Small 2018, 14, 1800665. [Google Scholar] [CrossRef]
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning: A comprehensive review. JAMA 2005, 294, 2342–2351. [Google Scholar] [CrossRef]
- Smallshaw, J.E.; Vitetta, E.S. Ricin Vaccine Development. In Ricin and Shiga Toxins: Pathogenesis, Immunity, Vaccines and Therapeutics; Mantis, N., Ed.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 259–272. ISBN 978-3-642-27470-1. [Google Scholar]
- Porter, A.; Phillips, G.; Smith, L.; Erwin-Cohen, R.; Tammariello, R.; Hale, M.; DaSilva, L. Evaluation of a ricin vaccine candidate (RVEc) for human toxicity using an in vitro vascular leak assay. Toxicon 2011, 58, 68–75. [Google Scholar] [CrossRef]
- Bascon, J.U. Vascular leak syndrome: A troublesome side effect of immunotherapy, Immunopharmacology, 39/3 (1998) 255. Immunopharmacology 1998, 39, 255–257. [Google Scholar]
- Vance, D.J.; Rong, Y.; Brey, R.N.; Mantis, N.J. Combination of Two Candidate Subunit Vaccine Antigens Elicits Protective Immunity to Ricin and Anthrax Toxin in Mice. Vaccine 2015, 33, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Vance, D.J.; Mantis, N.J. Progress and Challenges Associated with the Development of Ricin Toxin Subunit Vaccines. Expert Rev. Vaccines 2016, 15, 1213–1222. [Google Scholar] [CrossRef]
- Wahome, N.; Sully, E.; Singer, C.; Thomas, J.C.; Hu, L.; Joshi, S.B.; Volkin, D.B.; Fang, J.; Karanicolas, J.; Jacobs, D.J.; et al. Novel Ricin Subunit Antigens with Enhanced Capacity to Elicit Toxin-Neutralizing Antibody Responses in Mice. J. Pharm. Sci. 2016, 105, 1603–1613. [Google Scholar] [CrossRef]
- Sully, E.K.; Whaley, K.J.; Bohorova, N.; Bohorov, O.; Goodman, C.; Kim, D.H.; Pauly, M.H.; Velasco, J.; Hiatt, E.; Morton, J.; et al. Chimeric plantibody passively protects mice against aerosolized ricin challenge. Clin. Vaccine Immunol. 2014, 21, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Yermakova, A.; Klokk, T.I.; Cole, R.; Sandvig, K.; Mantis, N.J. Antibody-Mediated Inhibition of Ricin Toxin Retrograde Transport. mBio 2014, 5, e00995-13. [Google Scholar] [CrossRef] [PubMed]
- Herrera, C.; Vance, D.J.; Eisele, L.E.; Shoemaker, C.B.; Mantis, N.J. Differential neutralizing activities of a single domain camelid antibody (VHH) specific for ricin toxin’s binding subunit (RTB). PLoS ONE 2014, 9, e99788. [Google Scholar] [CrossRef]
- Vance, D.J.; Tremblay, J.M.; Rong, Y.; Angalakurthi, S.K.; Volkin, D.B.; Middaugh, C.R.; Weis, D.D.; Shoemaker, C.B.; Mantis, N.J. High-Resolution Epitope Positioning of a Large Collection of Neutralizing and Nonneutralizing Single-Domain Antibodies on the Enzymatic and Binding Subunits of Ricin Toxin. Clin. Vaccine Immunol. 2017, 24. [Google Scholar] [CrossRef]
- Poon, A.Y.; Vance, D.J.; Rong, Y.; Ehrbar, D.; Mantis, N.J. A Supercluster of Neutralizing Epitopes at the Interface of Ricin’s Enzymatic (RTA) and Binding (RTB) Subunits. Toxins 2017, 9, 378. [Google Scholar] [CrossRef]
- O’Hara, J.M.; Mantis, N.J. Neutralizing Monoclonal Antibodies against Ricin’s Enzymatic Subunit Interfere with Protein Disulfide Isomerase-Mediated Reduction of Ricin Holotoxin In Vitro. J. Immunol. Methods 2013, 395, 71–78. [Google Scholar] [CrossRef]
- Pincus, S.; Das, A.; Song, K.; Maresh, G.; Corti, M.; Berry, J. Role of Fc in Antibody-Mediated Protection from Ricin Toxin. Toxins 2014, 6, 1512–1525. [Google Scholar] [CrossRef]
- Yermakova, A.; Mantis, N.J. Protective Immunity to Ricin Toxin Conferred by Antibodies against the Toxin’s Binding Subunit (RTB). Vaccine 2011, 29, 7925–7935. [Google Scholar] [CrossRef] [PubMed]
- Yermakova, A.; Mantis, N.J. Neutralizing activity and protective immunity to ricin toxin conferred by B subunit (RTB)-specific Fab fragments. Toxicon 2013, 72, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yermakova, A.; Vance, D.J.; Mantis, N.J. Sub-domains of ricin’s B subunit as targets of toxin neutralizing and non-neutralizing monoclonal antibodies. PLoS ONE 2012, 7, e44317. [Google Scholar] [CrossRef]
- Cherubin, P.; Quiñones, B.; Teter, K. Cellular recovery from exposure to sub-optimal concentrations of AB toxins that inhibit protein synthesis. Sci. Rep. 2018, 8, 2494. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-Y.; Tserng, K.-Y.; Chen, C.-C.; Lin, L.-T.; Tung, T.-C. Abrin and Ricin: New Anti-tumour Substances. Nature 1970, 227, 292. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Chang, Y.C.; Huang, L.Y.; Tung, T.C. The cytotoxic effects of abrin and ricin on Ehrlich ascites tumor cells. Toxicon 1973, 11, 379–381. [Google Scholar] [CrossRef]
- Fodstad, O.; Kvalheim, G.; Godal, A.; Lotsberg, J.; Aamdal, S.; Høst, H.; Pihl, A. Phase I study of the plant protein ricin. Cancer Res. 1984, 44, 862–865. [Google Scholar]
- Davies, M.P.A.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar] [CrossRef]
- Koong, A.C.; Chauhan, V.; Romero-Ramirez, L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol. Ther. 2006, 5, 756–759. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sowa-Rogozińska, N.; Sominka, H.; Nowakowska-Gołacka, J.; Sandvig, K.; Słomińska-Wojewódzka, M. Intracellular Transport and Cytotoxicity of the Protein Toxin Ricin. Toxins 2019, 11, 350. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11060350
Sowa-Rogozińska N, Sominka H, Nowakowska-Gołacka J, Sandvig K, Słomińska-Wojewódzka M. Intracellular Transport and Cytotoxicity of the Protein Toxin Ricin. Toxins. 2019; 11(6):350. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11060350
Chicago/Turabian StyleSowa-Rogozińska, Natalia, Hanna Sominka, Jowita Nowakowska-Gołacka, Kirsten Sandvig, and Monika Słomińska-Wojewódzka. 2019. "Intracellular Transport and Cytotoxicity of the Protein Toxin Ricin" Toxins 11, no. 6: 350. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11060350