P-Cresylsulfate, the Protein-Bound Uremic Toxin, Increased Endothelial Permeability Partly Mediated by Src-Induced Phosphorylation of VE-Cadherin


Abstract
:1. Introduction
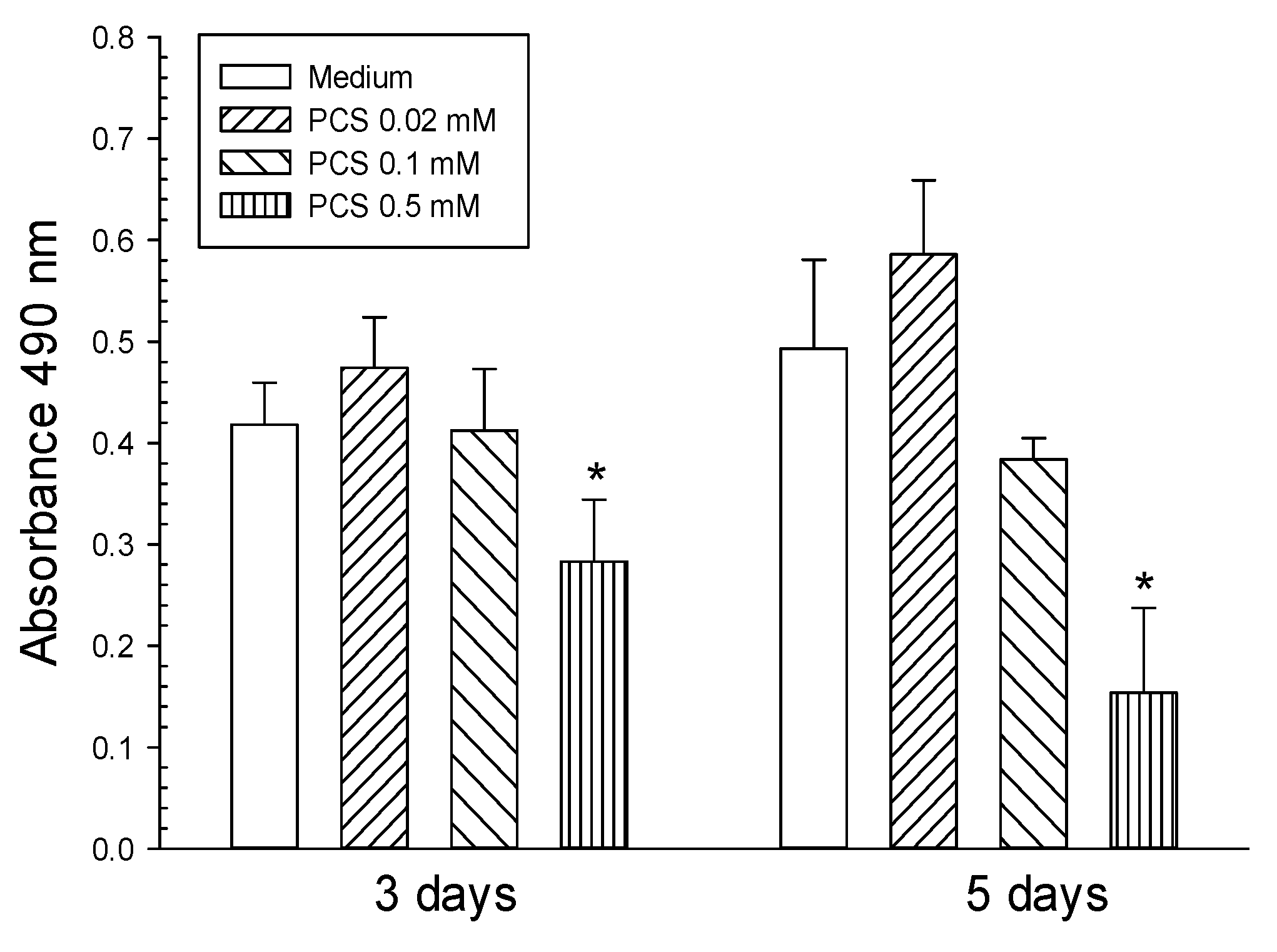
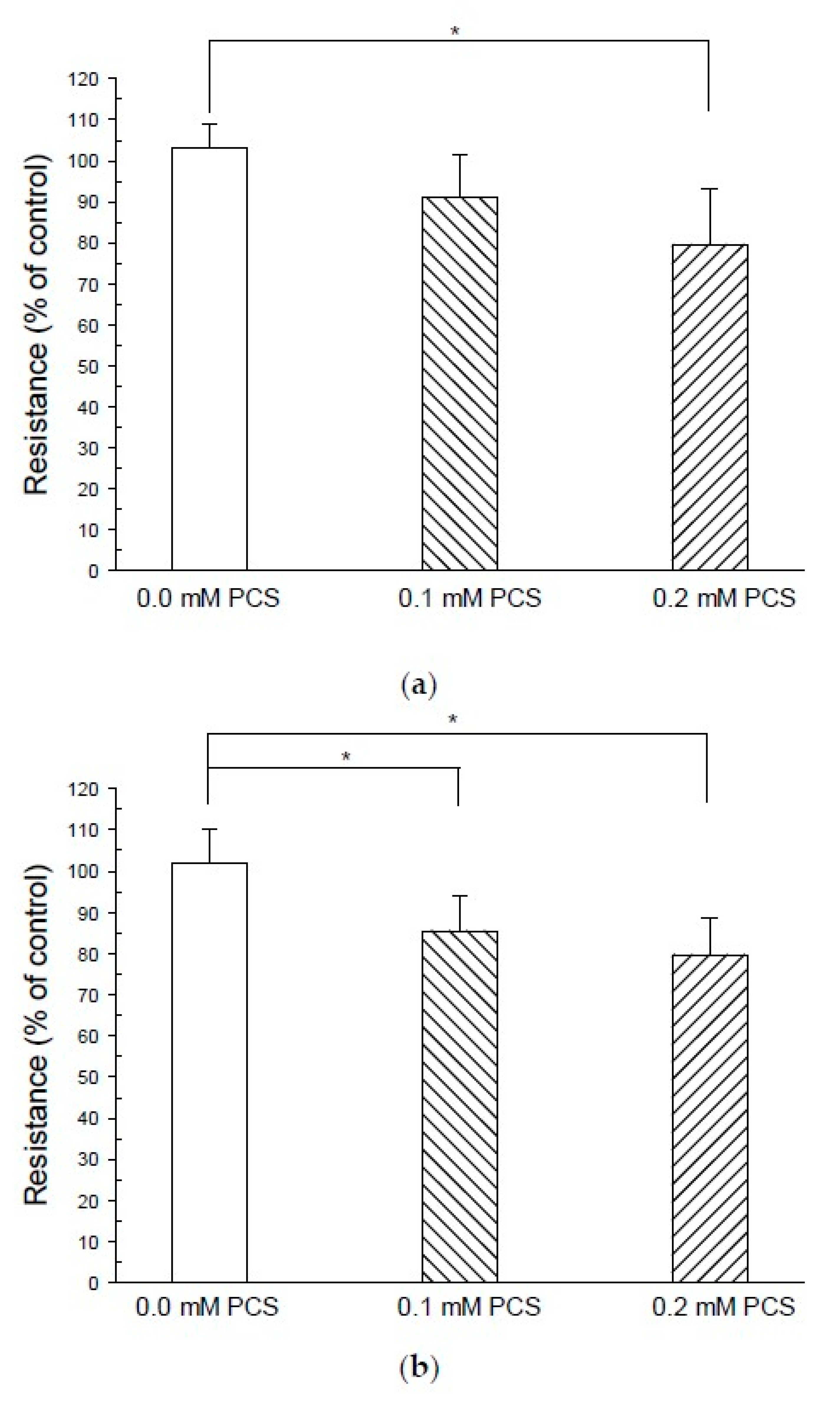
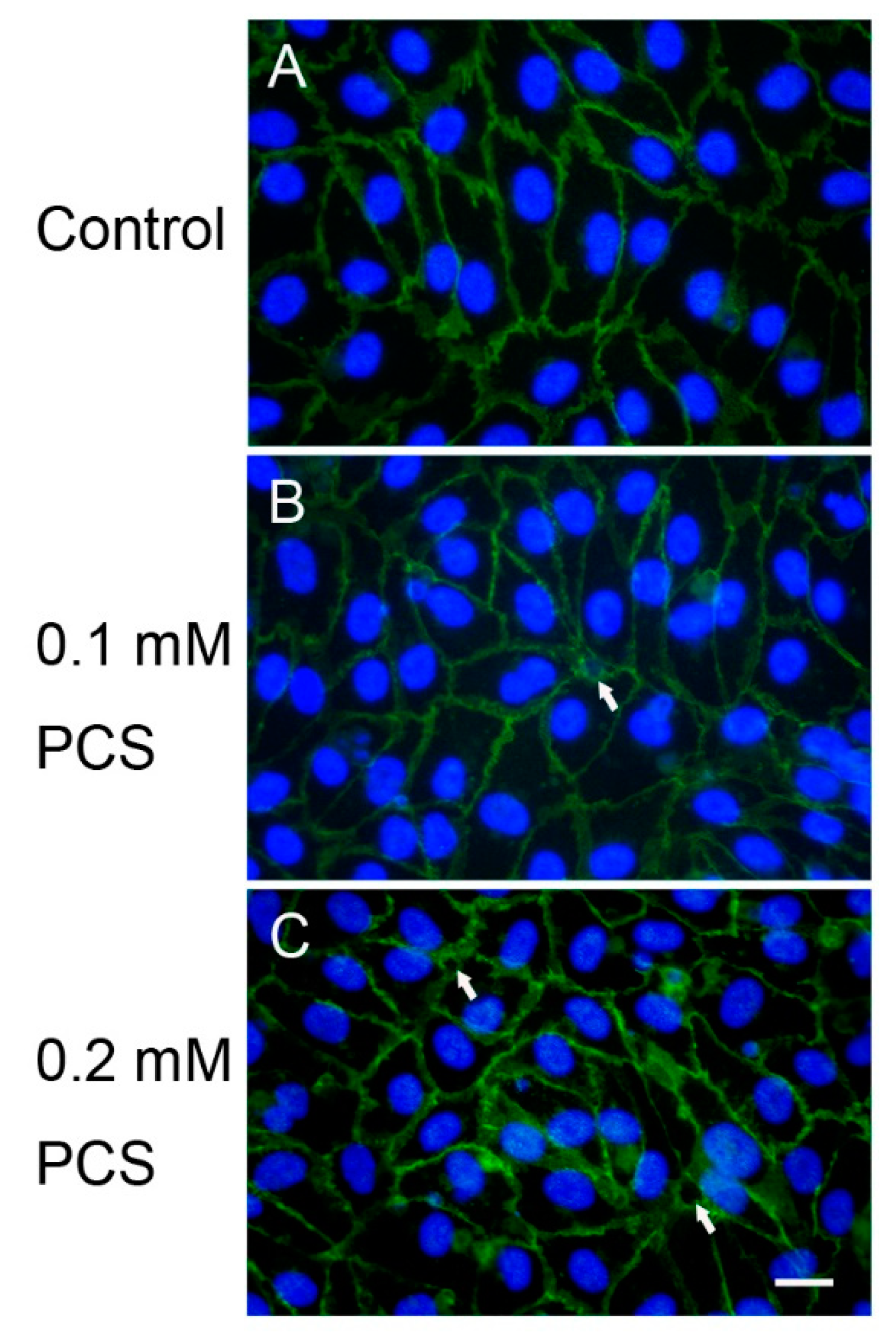
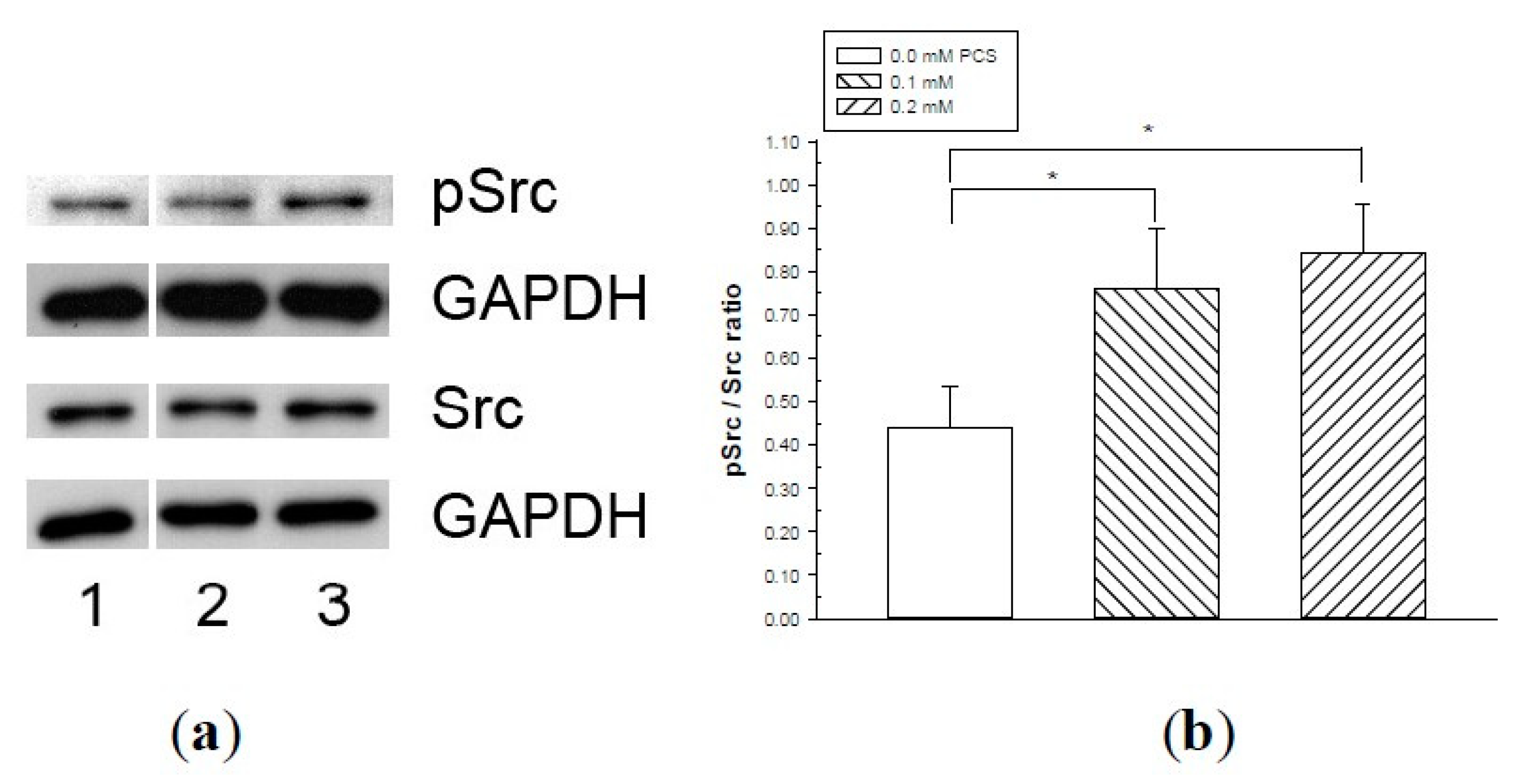
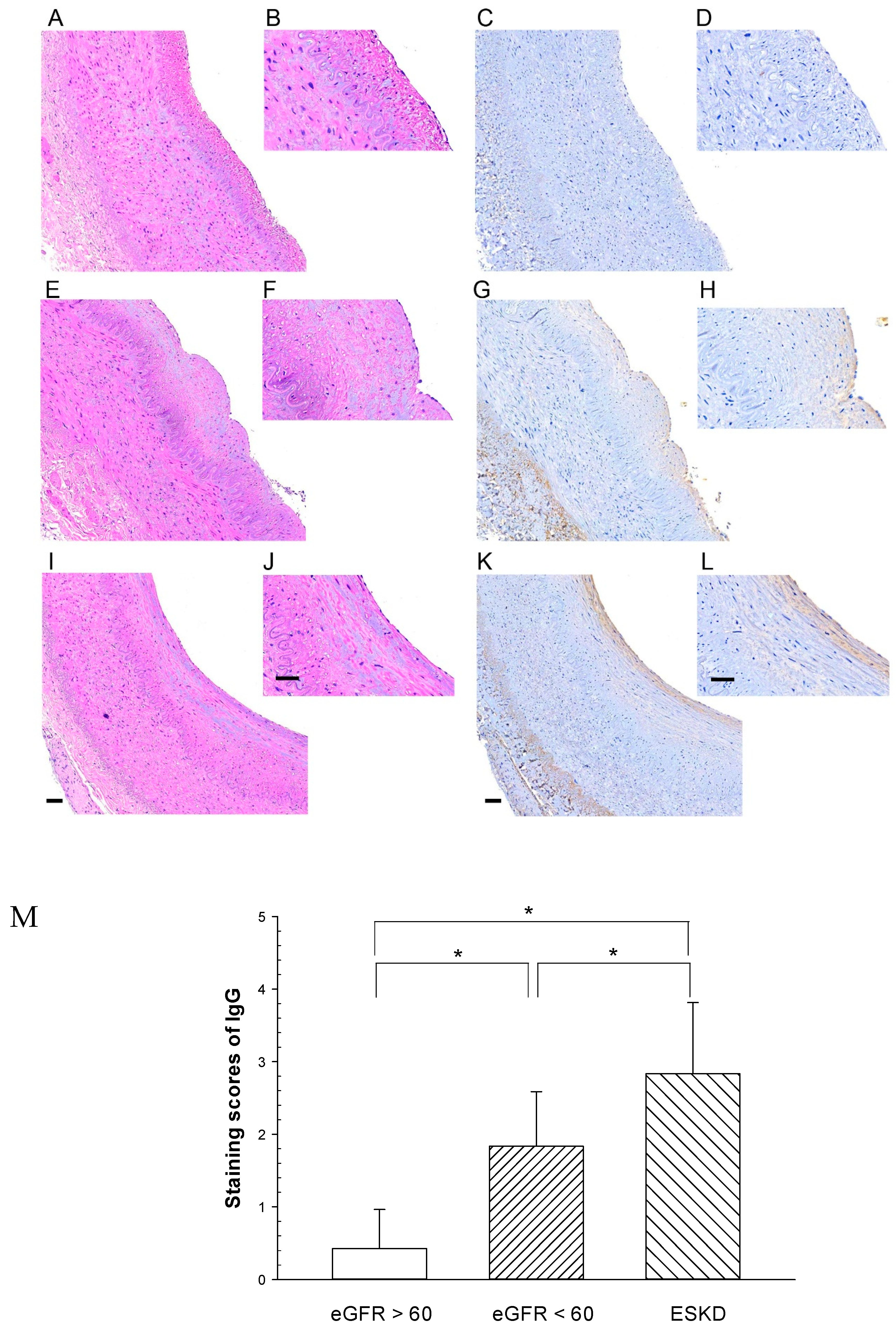
2. Results
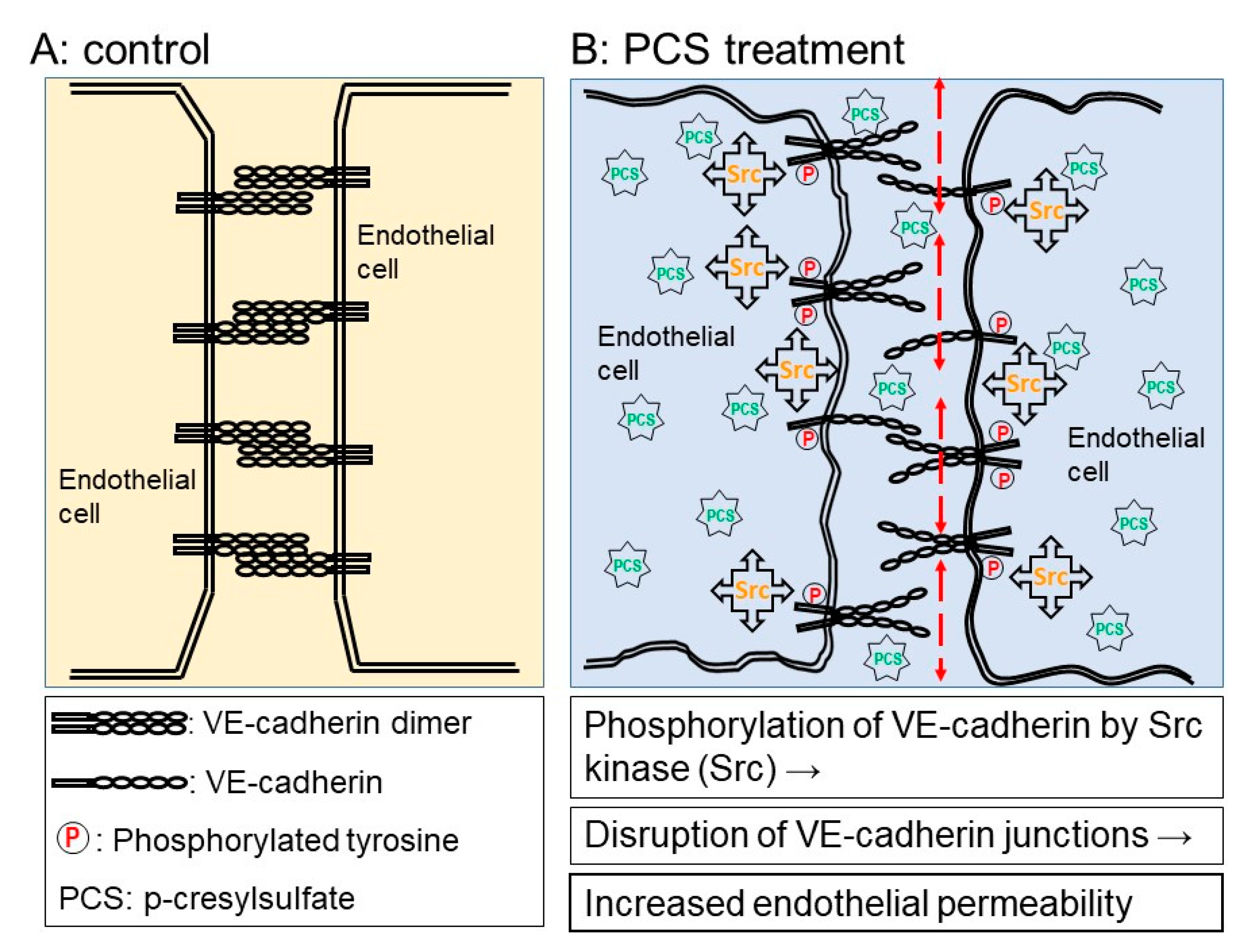
3. Discussion
4. Materials and Methods
4.1. Synthesis of P-Cresylsulfate (PCS)
4.2. Cell Culture and Cell Viability after PCS Treatment
4.3. TEER
4.4. Immunofluorescence Microscopy
4.5. Western Blotting
4.6. Microscopic Evaluation of the Endothelial Barrier of the Renal Artery of CKD Patients
4.7. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martinez, A.W.; Recht, N.S.; Hostetter, T.H.; Meyer, T.W. Removal of P-cresol sulfate by hemodialysis. J. Am. Soc. Nephrol. 2005, 16, 3430–3436. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T. Role of indoxyl sulfate in the progression of chronic kidney disease and cardiovascular disease: Experimental and clinical effects of oral sorbent AST-120. Ther. Apher. Dial. 2011, 15, 120–124. [Google Scholar] [CrossRef]
- Meijers, B.K.; Evenepoel, P. The gut-kidney axis: Indoxyl sulfate, p-cresyl sulfate and CKD progression. Nephrol. Dial. Transplant. 2011, 26, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.J.; Yen, C.H.; Wu, I.W.; Hsu, K.H.; Chen, C.K.; Sun, C.Y.; Chou, C.C.; Chen, C.Y.; Tsai, C.J.; Wu, M.S.; et al. The association of uremic toxins and inflammation in hemodialysis patients. PLoS ONE 2014, 9, e102691. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.J.; Ni, J.W.; Ding, F.H.; Fang, Y.H.; Wang, X.Q.; Wang, H.B.; Chen, X.N.; Chen, N.; Zhan, W.W.; Lu, L.; et al. p-Cresyl sulfate is associated with carotid arteriosclerosis in hemodialysis patients and promotes atherogenesis in apoE-/-mice. Kidney Int. 2016, 89, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef]
- Lee, C.T.; Hsu, C.Y.; Tain, Y.L.; Ng, H.Y.; Cheng, B.C.; Yang, C.C.; Wu, C.H.; Chiou, T.T.; Lee, Y.T.; Liao, S.C. Effects of AST-120 on blood concentrations of protein-bound uremic toxins and biomarkers of cardiovascular risk in chronic dialysis patients. Blood Purif. 2014, 37, 76–83. [Google Scholar] [CrossRef]
- Lin, C.J.; Chuang, C.K.; Jayakumar, T.; Liu, H.L.; Pan, C.F.; Wang, T.J.; Chen, H.H.; Wu, C.J. Serum p-cresyl sulfate predicts cardiovascular disease and mortality in elderly hemodialysis patients. Arch. Med. Sci. 2013, 9, 662–668. [Google Scholar] [CrossRef]
- Meijers, B.K.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef]
- Moradi, H.; Sica, D.A.; Kalantar-Zadeh, K. Cardiovascular burden associated with uremic toxins in patients with chronic kidney disease. Am. J. Nephrol. 2013, 38, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.P.; Lu, L.F.; Yu, T.H.; Hung, W.C.; Chiu, C.A.; Chung, F.M.; Yeh, L.R.; Chen, H.J.; Lee, Y.J.; Houng, J.Y. Serum levels of total p-cresylsulphate are associated with angiographic coronary atherosclerosis severity in stable angina patients with early stage of renal failure. Atherosclerosis 2010, 211, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients--a prospective cohort study. Nephrol. Dial. Transplant. 2012, 27, 1169–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravi, L.; Dejana, E.; Lampugnani, M.G. VE-cadherin at a glance. Cell Tissue Res. 2014, 355, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Shen, Q.; Pivetti, C.D.; Lee, E.S.; Wu, M.H.; Yuan, S.Y. Molecular mechanisms of endothelial hyperpermeability: Implications in inflammation. Expert Rev. Mol. Med. 2009, 11, e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Park-Windhol, C.; D’Amore, P.A. Disorders of Vascular Permeability. Annu. Rev. Pathol. 2016, 11, 251–281. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, E.; Mehta, D.; Minshall, R.; Malik, A.B. Regulation of endothelial junctional permeability. Ann. N. Y. Acad. Sci. 2008, 1123, 134–145. [Google Scholar] [CrossRef]
- Benn, A.; Bredow, C.; Casanova, I.; Vukicevic, S.; Knaus, P. VE-cadherin facilitates BMP-induced endothelial cell permeability and signaling. J. Cell Sci. 2016, 129, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E.; Tournier-Lasserve, E.; Weinstein, B.M. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev. Cell 2009, 16, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Hahn, C.S.; Scott, D.W.; Xu, X.; Roda, M.A.; Payne, G.A.; Wells, J.M.; Viera, L.; Winstead, C.J.; Bratcher, P.; Sparidans, R.W.; et al. The matrikine N-alpha-PGP couples extracellular matrix fragmentation to endothelial permeability. Sci. Adv. 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Monaghan-Benson, E.; Burridge, K. VE-cadherin status as an indicator of microvascular permeability. Methods Mol. Biol. 2013, 1046, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N.; Verin, A.; Catravas, J.D. Regulation of pulmonary endothelial barrier function by kinases. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L832–L845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.P.; Park, S.I.; Kopetz, S.; Gallick, G.E. Src family kinases as mediators of endothelial permeability: Effects on inflammation and metastasis. Cell Tissue Res. 2009, 335, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambeng, N.; Wallez, Y.; Rampon, C.; Cand, F.; Christe, G.; Gulino-Debrac, D.; Vilgrain, I.; Huber, P. Vascular endothelial-cadherin tyrosine phosphorylation in angiogenic and quiescent adult tissues. Circ. Res. 2005, 96, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Sukriti, S.; Tauseef, M.; Yazbeck, P.; Mehta, D. Mechanisms regulating endothelial permeability. Pulm. Circ. 2014, 4, 535–551. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, C.P.; Yu, T.H.; Tai, P.Y.; Liang, S.S.; Hung, W.C.; Wu, C.C.; Huang, S.H.; Lee, Y.J.; Chen, S.C. Protein-bounded uremic toxin p-cresylsulfate induces vascular permeability alternations. Histochem. Cell Biol. 2018, 149, 607–617. [Google Scholar] [CrossRef]
- Campbell, E.J.; McDuff, E.; Tatarov, O.; Tovey, S.; Brunton, V.; Cooke, T.G.; Edwards, J. Phosphorylated c-Src in the nucleus is associated with improved patient outcome in ER-positive breast cancer. Br. J. Cancer 2008, 99, 1769–1774. [Google Scholar] [CrossRef] [Green Version]
- Im, J.E.; Song, S.H.; Suh, W. Src tyrosine kinase regulates the stem cell factor-induced breakdown of the blood-retinal barrier. Mol. Vis. 2016, 22, 1213–1220. [Google Scholar]
- Zetser, A.; Bashenko, Y.; Edovitsky, E.; Levy-Adam, F.; Vlodavsky, I.; Ilan, N. Heparanase induces vascular endothelial growth factor expression: Correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006, 66, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.K.; Bammens, B.; De Moor, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. 2008, 73, 1174–1180. [Google Scholar] [CrossRef] [Green Version]
- Birukova, A.A.; Shah, A.S.; Tian, Y.; Gawlak, G.; Sarich, N.; Birukov, K.G. Selective Role of Vinculin in Contractile Mechanisms of Endothelial Permeability. Am. J. Respir. Cell Mol. Biol. 2016, 55, 476–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Wu, M.H.; Lee, E.S.; Yuan, S.Y. A disintegrin and metalloproteinase 15 contributes to atherosclerosis by mediating endothelial barrier dysfunction via Src family kinase activity. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2444–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestweber, D.; Wessel, F.; Nottebaum, A.F. Similarities and differences in the regulation of leukocyte extravasation and vascular permeability. Semin. Immunopathol. 2014, 36, 177–192. [Google Scholar] [CrossRef]
- Wessel, F.; Winderlich, M.; Holm, M.; Frye, M.; Rivera-Galdos, R.; Vockel, M.; Linnepe, R.; Ipe, U.; Stadtmann, A.; Zarbock, A.; et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 2014, 15, 223–230. [Google Scholar] [CrossRef]
- Sidibe, A.; Imhof, B.A. VE-cadherin phosphorylation decides: Vascular permeability or diapedesis. Nat. Immunol. 2014, 15, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef]
- Watanabe, H.; Miyamoto, Y.; Enoki, Y.; Ishima, Y.; Kadowaki, D.; Kotani, S.; Nakajima, M.; Tanaka, M.; Matsushita, K.; Mori, Y.; et al. p-Cresyl sulfate, a uremic toxin, causes vascular endothelial and smooth muscle cell damages by inducing oxidative stress. Pharmacol. Res. Perspect. 2015, 3, e00092. [Google Scholar] [CrossRef]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.S.; Ding, H.C.; Lin, Y.T.; Syu, J.P.; Chen, Y.; Wang, S.M. Uremic toxin p-cresol induces disassembly of gap junctions of cardiomyocytes. Toxicology 2012, 302, 11–17. [Google Scholar] [CrossRef]
- Cerini, C.; Dou, L.; Anfosso, F.; Sabatier, F.; Moal, V.; Glorieux, G.; De Smet, R.; Vanholder, R.; Dignat-George, F.; Sampol, J.; et al. P-cresol, a uremic retention solute, alters the endothelial barrier function in vitro. Thromb. Haemost. 2004, 92, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.S.; Lin, Y.T.; Chen, Y.; Hung, K.Y.; Wang, S.M. Effects of indoxyl sulfate on adherens junctions of endothelial cells and the underlying signaling mechanism. J. Cell. Biochem. 2012, 113, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef] [PubMed]
- Aman, J.; Weijers, E.M.; van Nieuw Amerongen, G.P.; Malik, A.B.; van Hinsbergh, V.W. Using cultured endothelial cells to study endothelial barrier dysfunction: Challenges and opportunities. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L453–L466. [Google Scholar] [CrossRef] [PubMed]
- Ursoli Ferreira, F.; Eduardo Botelho Souza, L.; Hassibe Thome, C.; Tomazini Pinto, M.; Origassa, C.; Salustiano, S.; Marcel Faca, V.; Olsen Camara, N.; Kashima, S.; Tadeu Covas, D. Endothelial Cells Tissue-Specific Origins Affects Their Responsiveness to TGF-beta2 during Endothelial-to-Mesenchymal Transition. Int. J. Mol. Sci. 2019, 20, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef] [Green Version]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Buricchi, F.; Raugei, G.; Ramponi, G.; Chiarugi, P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 2005, 25, 6391–6403. [Google Scholar] [CrossRef] [Green Version]
- Hoshi, Y.; Uchida, Y.; Tachikawa, M.; Ohtsuki, S.; Couraud, P.O.; Suzuki, T.; Terasaki, T. Oxidative stress-induced activation of Abl and Src kinases rapidly induces P-glycoprotein internalization via phosphorylation of caveolin-1 on tyrosine-14, decreasing cortisol efflux at the blood-brain barrier. J. Cereb. Blood Flow Metab. 2019. [Google Scholar] [CrossRef]
- Joris, I.; Cuenoud, H.F.; Doern, G.V.; Underwood, J.M.; Majno, G. Capillary leakage in inflammation. A study by vascular labeling. Am. J. Pathol. 1990, 137, 1353–1363. [Google Scholar]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Goto, S.; Fukagawa, M. Role of Uremic Toxins for Kidney, Cardiovascular, and Bone Dysfunction. Toxins 2018, 10, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirase, T.; Node, K. Endothelial dysfunction as a cellular mechanism for vascular failure. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H499–H505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.C.; Liu, C.C.; Huang, S.Y.; Chiou, S.J. Vascular hyperpermeability in response to inflammatory mustard oil is mediated by Rho kinase in mice systemically exposed to arsenic. Microvasc. Res. 2011, 82, 182–189. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Group 1 (n = 7) | Group 2 (n = 7) | Group 3 (n = 7) | p Value |
|---|---|---|---|---|
| Age (years) | 69.7 ± 12.6 | 76.7 ± 5.1 | 62.1 ± 11.4 | 0.011 |
| eGFR, mL/min/1.73 m2 | 73.4 ± 10.5 | 33.9 ± 15.9 | 6.4 ± 2.5 | *<0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-C.; Huang, S.-Y.; Wu, C.-C.; Hsu, C.-F. P-Cresylsulfate, the Protein-Bound Uremic Toxin, Increased Endothelial Permeability Partly Mediated by Src-Induced Phosphorylation of VE-Cadherin. Toxins 2020, 12, 62. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12020062
Chen S-C, Huang S-Y, Wu C-C, Hsu C-F. P-Cresylsulfate, the Protein-Bound Uremic Toxin, Increased Endothelial Permeability Partly Mediated by Src-Induced Phosphorylation of VE-Cadherin. Toxins. 2020; 12(2):62. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12020062
Chicago/Turabian StyleChen, Shih-Chieh, Shin-Yin Huang, Chia-Chun Wu, and Chiung-Fang Hsu. 2020. "P-Cresylsulfate, the Protein-Bound Uremic Toxin, Increased Endothelial Permeability Partly Mediated by Src-Induced Phosphorylation of VE-Cadherin" Toxins 12, no. 2: 62. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12020062