1. Introduction
The bio-accumulation of marine biotoxins produced by phytoplankton in filter-feeding invertebrates and finfish not only poses significant health threats to consumers but also has detrimental effects on the economies of nations highly dependent on seafood consumption for their subsistence [
1,
2,
3]. Among these potent marine biotoxins are neurotoxins acting on the voltage gated sodium channels (VGSCs) of excitable cells, namely VGSC activators such as brevetoxins (PbTxs) and ciguatoxins (CTXs), and VGSC inhibitors such as saxitoxins (STXs) and tetrodotoxins (TTXs) [
1,
2]. Detection and quantification of these groups of marine toxins remain highly challenging due to the wide range of congeners present in trace amounts in contaminated biological matrices [
4]. In addition, an official reference method is still lacking for most of these toxins due to the poor availability of certified reference standards [
5]. Although the mouse bioassay has been recommended by the European Food Safety Authority (EFSA) for years until 2015, the European Union now strongly advocates the use of analytical techniques based on liquid chromatography coupled with mass spectrometry in tandem (LC-MS/MS) and high performance liquid chromatography with fluorescence detection (HPLC-FLD) for the detection of lipophilic and hydrophilic toxins, respectively [
6,
7,
8,
9,
10]. However, the need for alternative high throughput methods for toxin screening and quantification purposes remains [
8,
11]. Among the readily available and most widely used in vitro methods, the functional assay known as the cell-based assay (CBA) that uses a neuroblastoma (N2a) cell line appears as the most promising one [
2,
12].
Based on the mode of action of VGSC toxins, the CBA-N2a was initially developed to detect two VGSC inhibitors, STXs and TTXs [
13]. Those marine toxins have no cytotoxic effect on N2a cells and their detection requires the addition of the sodium/potassium (Na
+/K
+) ATPase pump blocker ouabain (O), together with the sodium-channel activator veratridine (V), which induces permanent activation of the VGSCs [
14]. Under this O/V treatment (OV
+ conditions), sodium influx resulted in the cellular swelling and subsequent death of N2a cells [
13]. Following the addition of STX or TTX, an increase of cell viability is observed as the two inhibitors counteract the effect of ouabain and veratridine. At that time, viability measure was achieved by a visual estimation of the morphological changes of N2a cells and the enumeration of viable cells under an inverted microscope. This assay performed in a 96-well microplate format has been applied to the detection of Paralytic Shellfish Poisoning (PSP) toxin standards [
13] and to assess the presence of PSP toxins in biological extracts [
15,
16]. Standardization was also achieved using a certified STX standard for the detection of PSP toxins in shellfish and dinoflagellate cell extracts using automated endpoint determination with final readout based on crystal violet staining [
17]. Further, this CBA-N2a was applied to detect VGSC inhibitors in marine bacterial supernatants using neutral red for final estimation of toxicity [
18] and was subsequently extended to the detection of PbTxs and CTXs [
19,
20]. Contrary to VGSC inhibitors, these two VGSC activators induce a decrease in N2a cell viability in OV
+ conditions [
19,
20], whereas no cytotoxic effect is observed in the absence of O/V treatment [
21]. These same authors also introduced a more workable measure of cell viability using the 3-(4,5-dimethyl-2-thiazol)-2,5-diphenyl-2H-tetrazolium bromide (methylthiazolyldiphenyl-tetrazolium bromide) colorimetric assay (known as MTT assay), as modified from previous protocols aiming at assessing either cellular growth and survival [
22] or chemosensitivity in established cell lines [
23]. The MTT assay is widely used to measure cell viability and proliferation [
24,
25,
26,
27], and is based on the capacity of mitochondrial dehydrogenase enzymes present in living cells to reduce tetrazolium salts into an insoluble purple formazan product [
28] localized in lipids droplets [
29]. The readout step was first established using dimethyl-sulfoxide (DMSO) to dissolve formazan products [
23], the amount of the formazan produced is directly linked to the number of viable cells remaining at the end of the assay. This estimation is achieved by means of an automated readout. Two patents describing an improved and simplified CBA-N2a were eventually filed, leading to great expectations among the scientific community that this bioassay could soon be routinely used to detect both activators and inhibitors of sodium channels [
30,
31]. However, neither commercial kit nor detailed protocol is available to date.
Briefly, the CBA-N2a originally set by Manger et al. [
19,
20] consists of three major steps; (i) cell seeding of 100,000 cells/200 µL/well in a 96-well microplate left to grown for ≈ 24 h, (ii) O/V treatment followed by cell layer exposure to toxin standards and/or biological samples during 24 to 48 h, depending on the targeted toxin activity (final reaction volume of 230 µL/well) and (iii) N2a cell viability readout at 570 nm using the MTT colorimetric assay. Practically, the detection of VGSC activators and inhibitors by CBA-N2a requires 3 and 4 days, respectively.
Since 1993, the CBA-N2a has been widely used in a number of studies for the detection of VGSC toxins and related toxin families. However, a comprehensive review of the literature shows that the protocol initially defined by Manger et al. [
19,
20] has undergone numerous changes: for instance, (i) up to 13 different cell seeding densities have been tested, ranging from 10,000 to 250,000 cells/well [
32,
33,
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78,
79,
80,
81,
82,
83,
84,
85,
86,
87,
88] suggesting that cell confluence likely varied substantially between studies, especially when estimated by eye measurement; (ii) the culture medium established for cell layer implementation, which formerly used 10% of fetal bovine serum (FBS), was often reduced to 5% in many studies [
32,
33,
36,
37,
38,
39,
42,
46,
48,
54,
59,
69,
70,
74,
77,
78,
81,
82,
83,
85,
86,
89]; (iii) the confluence level reached by N2a cells after 24 h growth varied from non-confluent cells [
66] to >90% confluence [
48,
59,
63,
64,
65,
72,
73,
77,
78,
83] or even was not specified in some studies [
33,
34,
35,
36,
37,
38,
40,
41,
42,
43,
47,
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
61,
66,
67,
69,
70,
74,
75,
76,
79,
80,
81,
82,
85,
87,
88,
89,
90].
Other steps of the CBA-N2a have also been the subject of substantial modifications. For instance, although many studies used a 500/50 µM (1:10 ratio) for O/V treatment [
32,
33,
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
76,
89,
91,
92] as initially defined by Manger et al. [
19,
20,
30,
31], numerous changes in O/V concentrations [
54,
55,
56,
57,
59,
61,
63,
64,
65,
71,
72,
73,
74,
75,
77,
78,
79,
80,
81,
82,
83,
85,
87,
93,
94,
95], O/V ratios [
50,
51,
52,
69,
86] and reaction volumes [
32,
33,
35,
36,
49,
51,
53,
59,
61,
63,
64,
69,
73,
75,
77,
78,
79,
80,
84,
86,
87,
88,
89,
90] were further proposed. Interestingly, in many of these studies, the effects on cell viability resulting from all these modifications were rarely addressed but, when reported, showed incongruent results. For instance, O/V treatment at 500/50 µM was shown to induce highly variable effects between 20–92% [
34,
45,
68,
96]. Conversely, similar effects (10–56%) were reportedly obtained at lower O/V concentrations [
50,
86]. The most significant modification was the use of two different O/V treatments, i.e., 100/10 and 300/30 µM, in order to induce 20% and 80% of cell mortality, respectively, for a more reliable assessment of the effects of VGSC toxin activators and inhibitors on N2a cells [
83]. Several changes were also made to the final reaction volume (230 µL) at the last step of the CBA-N2a, i.e., cell exposure to toxin standards or biological samples, with volumes varying from 100 to 210 µL [
32,
33,
34,
35,
36,
45,
49,
51,
53,
59,
61,
63,
64,
69,
73,
75,
77,
78,
79,
80,
84,
86,
87,
88,
89,
90,
95]. Moreover, measuring cell viability by means of the MTT colorimetric assay is classically performed at 570 nm, however, different wavelengths were also tested that ranged from 490 to 595 nm [
33,
35,
36,
38,
39,
40,
41,
42,
45,
50,
56,
57,
61,
64,
67,
68,
69,
71,
75,
76,
79,
80,
84,
85,
87,
88,
90,
93,
97]. Finally, water-soluble tetrazolium reagents were sometimes preferred to the former water insoluble formazan product, with the use of WST-8 [
49,
60] or commercial kits such as CellTiter 96
® Aqueous One solution [
33,
38,
69,
86], Cell Counting Kit-8 [
47] or XTT Cell Proliferation Assay kit [
98].
This wide range of “in-house” methods described in the literature highlights the present lack of a consensus, standardized protocol for CBA-N2a, making any attempt to compare CBA data between assays and/or laboratories difficult or impossible. It also greatly hampers all current efforts to establish the CBA-N2a tool as a potential alternative reference method to LC-MS/MS. In this context, the present work aims at a comprehensive revisit of the CBA-N2a by evaluating the effects of six key parameters that are critical in obtaining reliable toxicity results, i.e., cell seeding densities, cell layer viability after 26 h growth, MTT incubation time, O/V treatment and solvent and matrix effects. To evaluate its robustness, the newly improved protocol thus established was further applied to the detection of toxins active on VGSC families (activators vs. inhibitors), and the estimation of CTX-like toxicity in fish samples of known ciguatoxic status.
3. Discussion
Having sensitive and specific detection methods available to protect consumers against poisoning risks due to seafood contaminated with marine biotoxins is a key component of food security monitoring programs worldwide [
1,
5]. However, in the absence of duly validated reference methods, many marine biotoxins are still unregulated [
5]. This is the case for instance for CTXs, neurotoxins active on VGSCs that are implicated in ciguatera poisoning [
5]. Among the many detection tests currently available for the detection of this group of compounds and, more widely, of toxins acting on voltage-gated sodium channels [
8,
99], the neuroblastoma cell-based assay or CBA-N2a appears as a very sensitive, specific functional assay [
3,
12]. However, the numerous protocols found in the literature highlight the lack of a consensus standardized protocol for CBA-N2a. It should be noted that the same observation applies to other analytical methods used for CTXs determination in biological samples such as LC-MS/MS and references therein, [
99,
100,
101]. In this context, the present work aimed at revisiting several key parameters of the CBA-N2a as a step towards the standardized and reliable detection of two groups of toxins frequently involved in toxic outbreaks, namely VGSC activators (e.g., CTXs, PbTxs) and inhibitors (e.g., STX, dc-STX).
3.1. Revisit of the CBA-N2a
Overall, six key parameters of the CBA-N2a were revisited.
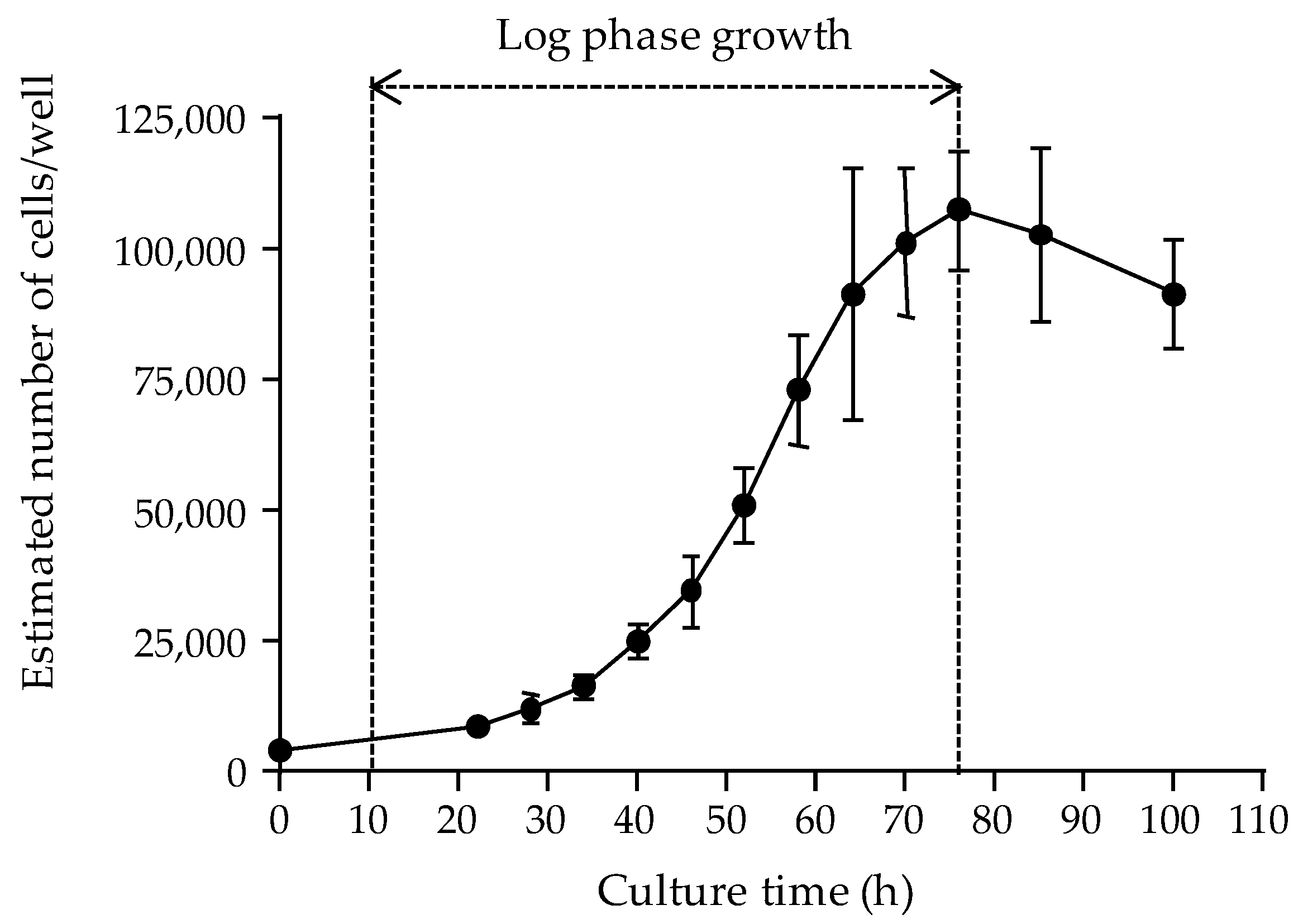
The first parameter was the cell layer density reached in microplate wells following a 22 to 26 h growth period at 37 °C, as assessed by measuring the absorbance values of the cell layer. The growth curve of the N2a cell line used in this study was established, demonstrating that the maximum number of cells that can be supported in 0.32 cm
2 wells could not exceed ≈ 100,000 cells. Our data also showed that seeding microplates at 50,000 cells/well allowed reaching this high cell density after ≈ 22 h of culture, with 100% confluence of cell layer in wells, consistent with results obtained in previous studies [
59,
77]. Obtaining maximum confluence with cells in late log growth phase ensures optimal absorbance data [
23]. In the literature, however, a cell layer confluence > 90% was reportedly reached using different cell seeding densities, e.g., 10,000 cells/well [
63], 30,000 cells/well [
64,
73] and 100,000 cells/well [
19,
20,
47,
48]. Some authors reported higher cell seeding densities of 250,000 cells/well [
57,
71], but such assay conditions do not appear physically or physiologically relevant. Actually, our results suggest that it is not the cell seeding density in itself that matters when seeking to achieve an optimal implementation of cell layer in culture microplates, and that this parameter should be adjusted to both the growth rate of the N2a cell line and the culture conditions used in the different laboratories. Therefore, growth experiments should be conducted in each laboratory in order to characterize the growth pattern of the cell line(s) in use.
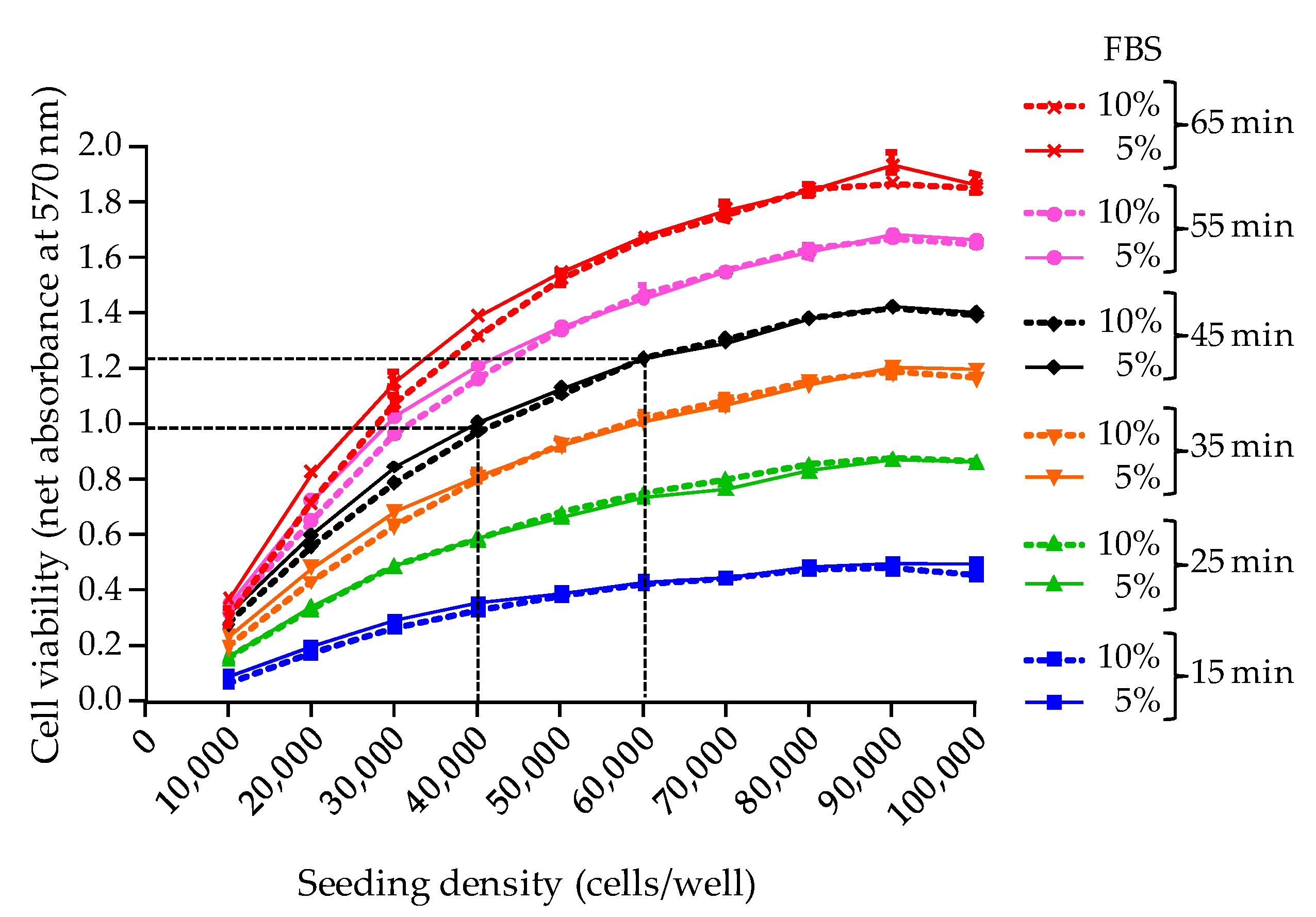
The second parameter examined in this study was the MTT incubation time for cell viability assessment [
19]. Here, the MTT assay was standardized using an optimal wavelength of 570 nm and an incubation time of 45 min, allowing to obtain reproducible absorbance values ≥ 1 when a maximum density of adherent cells is reached in wells after 26 h of growth, i.e., ≈ 100,000 cells/well. In all further experiments, this condition which serves as an indicator of the good health conditions of N2a cells before exposure to toxic extracts was referred to as the “initial cell viability”, and was assessed by implementing a RCV control microplate. Moreover, setting the initial cell viability to an absorbance value ≥ 1 (=baseline viability) allowed us to establish a full sigmoidal dose-response curve derived from testing an eight points serial dilution of the toxin standard/sample. Any subsequent drop in cell viability following O/V treatment and toxin exposure, could then be most likely attributed to the specific effect of toxin standard/biological sample, and not from non optimal culture conditions or oversensitivity of cells.
Regarding the selection of a wavelength at 570 nm, our results are in agreement with previous work showing that the peak absorbance of MTT formazan precipitate is classically observed at 562 nm with shoulders at 512 and 587 nm [
29]. Wavelengths within this range have been used in a number of studies, [
36,
42,
50,
64,
67,
87,
90], and were generally associated with MTT incubation time around 30 min. However, wavelengths outside this range were also tested, resulting in a weaker optical signal [
29] consistent with the fact that the use of lower [
45,
76,
84,
85] or higher wavelengths [
57,
61,
79,
93] often required longer MTT incubation times (>60 min).
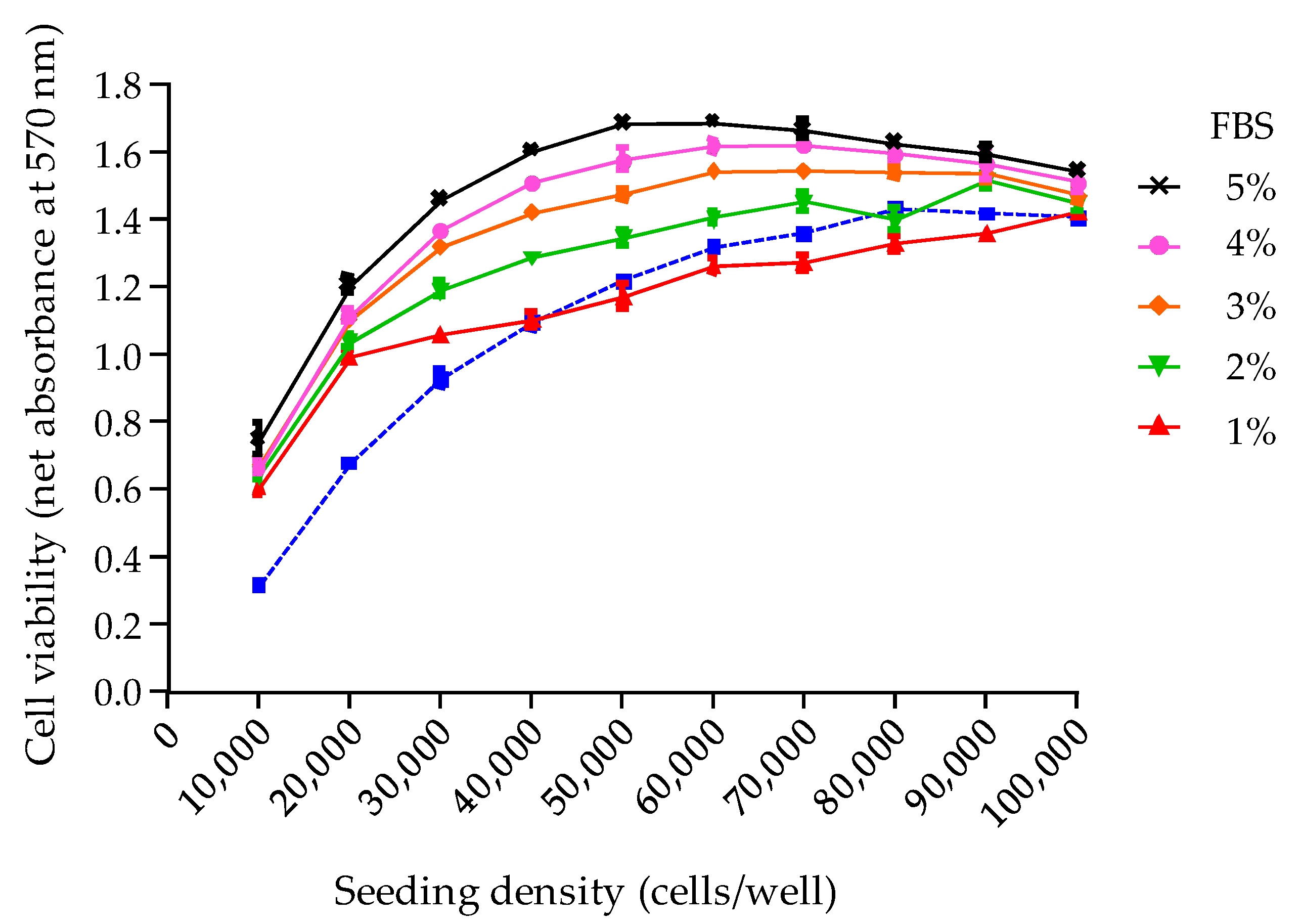
The third parameter analyzed was the final viability of N2a cells under OV
− conditions, following an additional culture period of 19 h, overnight. Indeed, the N2a growth curve previously established showed that a loss in N2a cell viability is likely to occur at this stage when a high cell density is reached. To prevent this, a complete renewal of the used growth medium with a 2% FBS culture medium was found sufficient to stabilize cell viability throughout the CBA-N2a, as evidenced by absorbance data that were consistently found ≥1 in COV
− control wells. This operation appears all the more relevant since MTT assay does not actually measure the number of viable cells or their growth, but the activity of an integrated set of enzyme linked to cell metabolism [
24]. The necessity of this medium renewal has been previously highlighted in many studies [
34,
49,
51,
56,
71,
79,
80,
87,
93] although the benefits of such a procedure were never clearly explained.
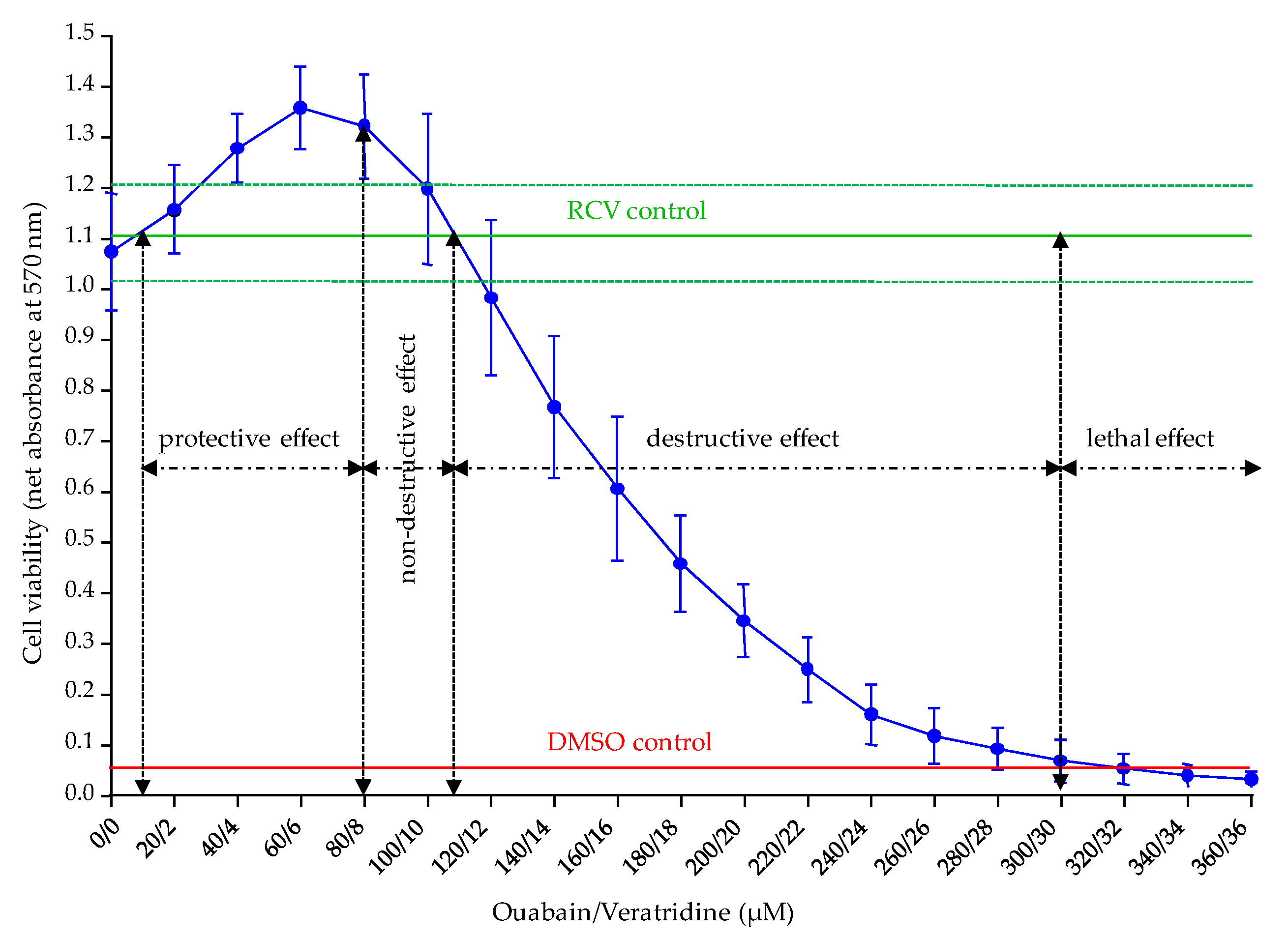
The fourth parameter revisited was the concentrations of Ouabain and Veratridine applied in O/V treatment. In this study, we were able to determine two distinct ranges of O/V concentrations to use depending on the group of toxins targeted in the CBA-N2a, i.e., VGSC activators and inhibitors. For the specific detection of VGSC activators in which a loss in cell viability is sought, non-destructive concentrations of O/V with respect to N2a cell viability ranging from 80/8 and 100/10 µM are recommended in order to maintain a final cell viability in COV
+ control close to the initial cell viability of RCV control (absorbance values ≥ 1). Conversely, for the specific detection of VGSC inhibitors in which a restoration of cell viability is sought, destructive concentrations of O/V ranging from 270/27 to 300/30 µM should be used in order to induce a residual cell viability in COV
+ control corresponding to approximately 10% (absorbance value of ≈ 0.1) of the initial cell viability measured in the RCV control. Moreover, the ranges of these two types of O/V treatments seem to induce constant effects on N2a cells although high passages were used and that these latter were distant from each other up to 420 passages. However, passage specific effects were not tested directly in this study, and may be the focus of future work. These findings are currently verified on the N2a cell line used in our laboratory at lower number of cell passages. The 500/50 µM O/V treatment originally proposed by Jellett [
17] to detect STX was in fact chosen to induce “a lysis of many of the cells in 24 h incubation time”. Such characteristics reinforce the idea that the CBA-N2a is a bioassay that is function-specific rather than toxin-specific [
102].
The use of 83/17 µM [
50] or 100/10 µM [
83] in O/V treatment has been reported in the literature for the detection of PbTxs, although a 10–20% reduction in cell viability was observed. Similarly, a 100/10 µM O/V treatment is often selected for the detection of CTXs in
Gambierdiscus spp. extracts [
56,
65,
79,
80,
93] and fish samples [
54,
55,
57,
71,
74,
80], although in many of these studies no information on whether a renewal of the growth medium was applied prior to O/V treatment or on the efficiency of O/V treatment was provided. Consistent with our results is the report of the use of a 300/30 µM O/V treatment in previous studies aiming at detecting STX, which resulted in an 80% reduction in cell viability [
83,
94]. Additionally, previous studies showed a reduction of cell viability of 40% and up to 69–92% without and following the renewal of the growth medium, respectively, under O/V treatment at 500/50 µM [
34,
96]. In the present study, an O/V treatment at 300/30 µM was sufficient to induce a 100% reduction in cell viability, suggesting that the renewal of the medium was beneficial to cell physiology, thus allowing to optimize the effect of O/V treatment. However, the extent to which reduction of FBS in the culture medium from 5% to 2% following medium renewal might have rendered the cells more sensitive to toxins remains to be tested in follow-up experiments. The direct addition of O/V treatment in old culture medium following cell layer implementation in wells [
19] should be avoided to ensure appropriate preservation of cell viability and further O/V treatment efficiency.
The fifth parameter examined was the concentration of solvents used to resuspend toxin standard solutions or dry extracts. This study showed that, for MeOH and DMSO, the highest final concentrations should be 0.5% to avoid non-specific cytotoxic effects on N2a cell viability, which is ensured by a dilution of at least 1:200 of the sample stock solutions. These possible solvent interferences were addressed in various ways in previous studies, including working at a final solvent concentration of 0.25–0.3% [
11,
87], performing an additional evaporation step before re-dissolution of toxins or biological samples in RPMI medium [
54,
74,
81,
91,
103], or adding 1% MeOH in controls wells to identify any cytotoxic effects [
64,
73] or 5% MeOH known to induce no more than 20% of cytotoxicity on cell lines [
53].
The sixth and last parameter was the maximum amount of extract to expose to avoid potential matrix effects (MCE) as compounds that often co-extract with the analyte(s) of interest can induce unspecific effects on N2a cell viability [
81]. In this study, this value was determined at 10,000 pg of dry extract/mL based on the LF90/10 dry extract weight (DEW) instead of the fresh tissue equivalent (FW) used to prepare these extracts, given the significant differences often observed in the DEW/FW ratio of biological samples. Interestingly, only one previous study has used this rationale and has calculated the MCE based on lipid extracts [
84]. It should be noted that MCE values are likely to vary according to the nature of tissue analyzed (e.g., flesh, viscera, fin, etc.), the different trends in lipid and water contents in tissue when sampled at different stages of the life history of a given fish species [
104]. Moreover, MCE also depends on the extraction protocols and extraction efficiency [
46,
57,
73,
105], as shown for CTXs for which no reference consensus extraction protocol is available as yet [
105]. Thus, until a standardized universal protocol is made available, researchers conducting similar studies should necessarily perform their own matrix assessments before applying CBA-N2a.
3.2. Performance of the Revisited CBA-N2a
The accuracy of the data provided by this revisited CBA-N2a, relies, in part, on the availability of five viability controls, i.e., RCV, COV
−, COV
+, QCOV
− and QCOV
+ that were established throughout the various stages of CBA-N2a in order to verify: (i) the initial (RCV control) and final viability of N2a cells (COV
− controls), (ii) the efficiency of O/V treatment (COV
+ controls) and (iii) the detection of the specific mode of action of VGSC activators vs. inhibitors (QCOV
− and QCOV
+ controls). If the use of percentages is recurrent in most studies to express cell viability results [
7,
8,
32,
33,
36,
37,
43,
46,
53,
59,
62,
63,
66,
67,
68,
70,
73,
74,
76,
81,
83,
85,
87,
88,
90,
91,
95,
97,
103,
106,
107,
108,
109,
110,
111,
112], the originality of our revisited CBA-N2a is to recommend the use of absorbance data by establishing a RCV control that characterizes the baseline viability of N2a cell layer prior to O/V treatment and toxin exposure. This RCV control serves as a reference from which all other viability controls as well as viability data of standard and samples depend. This RCV control could be considered as a common reference across all laboratories employing this method.
As a result of this comprehensive revisit of the CBA-N2a, a practical guide with a step-by-step protocol was defined (
Supplementary Materials), that is accessible to both supervised beginners and experienced users. It also provides useful decision limits when interpreting CBA-N2a data in the framework of routine CBA-N2a based monitoring programs.
In this study, the sensitivity of the CBA-N2a was characterized by EC
50 values found for P-CTX3C at 1.7 ± 0.35 pg/mL, consistent with previously published values of 1.3 ± 0.06 pg/mL [
46], 1.66 ± 0.16 pg/mL [
64,
73] and 1.44 ± 0.70 pg/mL [
77,
78]. However, quite different values can also be found in the literature, e.g., 0.57 ± 0.11 pg/mL [
90], 0.914 ± 0.127 pg/mL [
36] and 3.10 ± 0.76 pg/mL [
59]. These discrepancies can be attributed to different experimental conditions including different concentrations of O/V treatment [
73] or the use of MeOH as a sample vehicle for higher sensitivity [
90]. The EC
50 found for PbTx3 was 5.8 ± 0.9 ng/mL, which is lower than the previously published value of 65.60 ± 23 ng/mL [
37].
When comparing the mean EC
50 values of P-CTX3C to those obtained via other functional assays, they were found 350-fold lower than those estimated via the radioactive receptor binding assay (rRBA), i.e., 0.62± 0.16 ng/mL [
113] and 0.61 ± 0.01 ng/mL [
46], and the fluorescent RBA, i.e., 0.66 ± 0.16 ng/mL [
73], respectively. For PbTx3, EC
50 values were 2-fold higher than those derived from rRBA experiments (2.77 ± 1.09 ng/mL [
37] and 2.06 ± 0.16 ng/mL [
113]). These discrepancies observed between functional assays can be explained by the fact that the activity of these polyether toxins depends not only on their mode of action, and their affinity for specific sodium channel isoforms [
114], but also on their efficacy on other ion channels and references therein, [
37,
115,
116,
117]. As for the detection of STX and dc-STX, the comparison of CBA-N2a results obtained in this study with other functional assays was not possible since previous studies used different units.
For VGSC inhibitors, STX and dc-STX displayed typical dose-response curves with positive Hillslope in the presence of lethal 270/27 µM OV treatment giving EC
50 values of 3 ± 0.5 and 15.8 ± 3 ng/mL for STX and dc-STX, respectively. At the EC
20, this revisited CBA-N2a could specifically detect inhibition of VGSCs at 1 and 5 ng/mL for STX and dc-STX, respectively. This result is consistent with a detection limit established at 2 ng/mL for STXs by Manger et al. [
19].
This improved CBA-N2a was further applied to the detection of CTX-like toxicity in fish matrix, giving mean LOD and LOQ values of 0.031 ± 0.008 and 0.064 ± 0.016 ng P-CTX3C eq/g fish flesh, respectively. In order to compare these results with those obtained via analytical techniques such as LC-MS/MS data, this LOD value was further expressed in P-CTX1B equivalent, taking into account a toxicity equivalent factor (TEF) of 0.2 for P-CTX3C [
118]. A LOD value of 0.0062 ng P-CTX1B eq/g. was found below the recommended threshold of 0.01 ppb for P-CTX-1B [
118,
119], indicating that both CBA-N2a and LC-MS/MS share similar level of sensitivity for the detection of P-CTXs [
61,
100,
120,
121,
122]. However, chromatographic analyses remain unavoidable for the formal characterization of the different structural analogs (toxin congeners) present in biological samples.
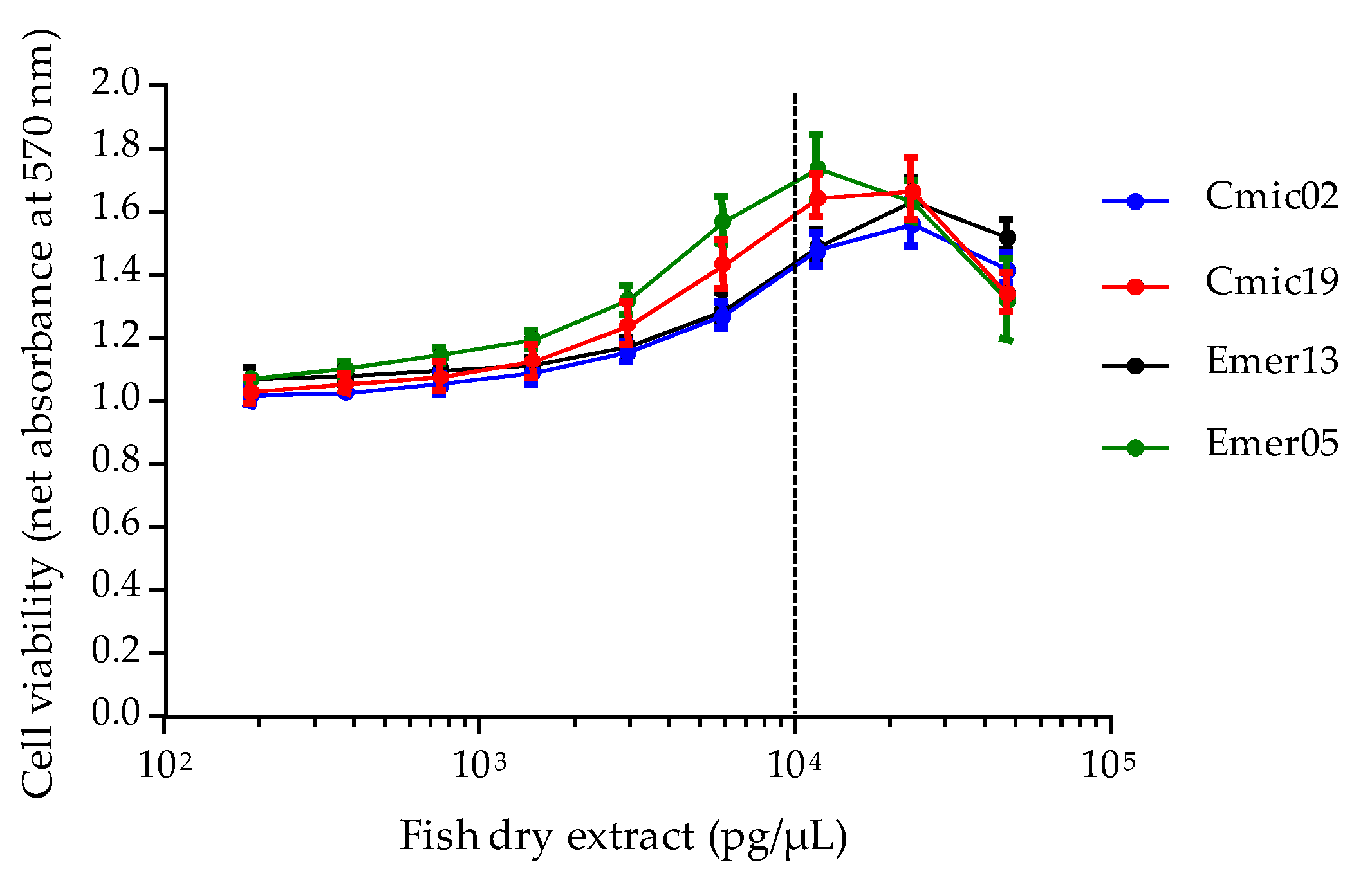
Based on EC
50 data, the toxin content in four fish samples of known ciguatoxic status was further checked via the CBA-N2a. Only two fish specimens, namely
Chlorurus microrhinos (Cmic19) and
Epinephelus merra (Emer05) were found to contain 6.66 ± 0.68 and 3.31 ± 0.35 ng P-CTX-3C eq/g fish flesh, respectively, corresponding to ≈ 1.34 and 0.65 ng P-CTX1B eq/g, which are 134- and 65- fold above the advisory level recommended by both the US FDA and EFSA [
118,
119]. The composite cytotoxicity detected in the Cmic19
Chlorurus microrhinos is in agreement with fRBA and LC-MS/MS analyses, which confirmed the presence of six distinct P-CTX congeners in this fish [
120]. Interestingly, these toxicity data also confirmed that the overall toxicity of herbivorous fish (parrot-fish) can sometimes surpass that of a carnivorous fish (grouper), although CTX toxin profiles occurring in individuals with different trophic habits can differ significantly owing to the biotransformation processes that occur along the food chain [
120,
123,
124]. In addition to biotransformation, several other traits can be responsible for the unique CTX toxin profiles of individual fish, including site-specificity, feeding behavior and ontogenetic dietary shifts [
61,
123,
125].
Several intra- and/or inter-assay comparisons of the revisited CBA-N2a were performed to assess the coefficients of variations (CVs) obtained for key parameters of CBA-N2a dose-response curves, and evaluate the repeatability and reproducibility of the method. Results indicate that for both VGSC activators and inhibitors, EC
50 values showed CVs ranging from 10% to 26%, which are considered as acceptable values for functional tests. These results are congruent with previous studies showing 3.5 to 25% and 5 to 24.7% of CBA-N2a variability for intra and inter-assay, respectively [
46,
80,
93,
126]. A variability of up to 30% is generally admitted for other functional tests [
127,
128,
129]. Additionally, no significant differences were found for EC
50 and EC
80 values of P-CTX3C and PbTx3 standards obtained from two non destructive O/V treatments run at distinct cell passages. Using different OV concentrations selected in the range of 80/8–100/10 µM and different numbers of cell passage seem to not impact the values of EC
50 and EC
80 in our study. For LOD and LOQ values, CVs below 29% were obtained vs. CV of 13% for composite toxicity estimates in the two ciguatoxic fishes.
3.3. Advantages of the Modified Protocol
An advantage of the CBA-N2a is necessary material and reagents, as well as basic laboratory equipments are readily (commercially) available.
Several opportunities to reduce reagents were identified at different steps of the test, e.g., using 5% instead of 10% FBS culture medium for the seeding of N2a cells, renewing culture medium with only 2% FBS growth medium as opposed to 5% or 10% FBS commonly used in many studies [
34,
49,
51,
56,
71,
79,
80,
87,
93], or working at lower O/V concentrations for O/V treatment than those originally proposed [
83]. Moreover, the direct addition of O/V solution into the renewed growth medium also contributes to a better homogeneity of O/V deposit for increased repeatability and reproducibility, and a reduction of variability sources by limiting the number of pipetting. Finally, this revisited protocol allowed the use of reduced amounts of expensive toxin standards with e.g., only 80 pg of P-CTX3C toxin standard required per microplate to establish a full dose-response curve.
Another advantage of this improved test is its high modularity with respect to the specific detection of a wide range of toxins acting on VGSCs, i.e., both VGSC activators and inhibitors, thanks to the definition of O/V treatment conditions allowing the visualization of a drop in cell viability vs. cell layer restoration, respectively. This protocol also offers the possibility to assay as many as 15 microplates in one experiment (in addition to the one used for RCV control) that can be used either for the qualitative screening of 120 distinct samples at a single concentration (
Figures S3 and S4), or the composite toxicity analyses of 14 samples and one toxin standard tested at eight distinct concentrations (
Figures S5 and S6), depending on the laboratory research goals. For instance, the high throughput screening approach would be more suitable in risk monitoring programs aiming at the random testing of a high number of wild fish specimens, e.g., to confirm the bioaccumulation of CTXs in the marine fauna and/or the emergence of CP in novel areas. The composite toxicity estimation would be preferred when confirmatory analyses in fish products are required, e.g., to confirm the diagnosis of CP in patients, or help link the CTX content in fish to the symptoms and/or severity of the poisoning. Alternatively, using a two-tiered approach in monitoring programs is also feasible, i.e., perform rapid screening tests on a whole batch of fish specimens, and then conduct composite toxicity estimations only on those specimens found suspect or positive.
3.4. Limitations and Gaps of the Present Study
Despite the significant improvements described in the present study, the effects of several parameters that were not tested here remain to be evaluated prior to the standardization and widespread use of this method by different laboratories. For example, parameters inherent to slight differences in N2a cell lines across laboratories due e.g., to mycoplasma infection of cell cultures [
130,
131], cell passage numbers [
132,
133], desensitization treatment [
134] or the use of different mammalian cell lines [
53,
66,
83,
135] should be considered in follow-up studies, as they are likely to affect cell growth rates and cell responses to toxins in this functional assay. Additional studies about the effects of reagents, media exchange, media renewal, etc. are also needed. Likewise, the inclusion of a limited number of fish in the matrix interference study presently limits the application of the method for general detection of VGSC toxins in other matrices (e.g., screening of PbTxs and STXs in shellfish samples). Another major issue is the current lack of a duly validated standardized extraction protocol for the detection of CTXs [
100,
105]. To this respect, the MCE value determined here must not be regarded as a universal dose as MCE closely depends not only on the extraction protocol used but also on the nature of the extract being tested [
105]. Therefore, laboratories would need to perform their own matrix assessments prior to CBA-N2a studies.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}