Molecular Tracing of SARS-CoV-2 in Italy in the First Three Months of the Epidemic


 ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Methods
2.2. Sequence Data Sets
2.3. Genetic Distance, Recombination, and Selection Pressure Analyses
2.4. Phylogenetic and Phylodynamic Analyses
2.5. Birth-Death Skyline Estimates of the Effective Reproductive Number (Re)
3. Results
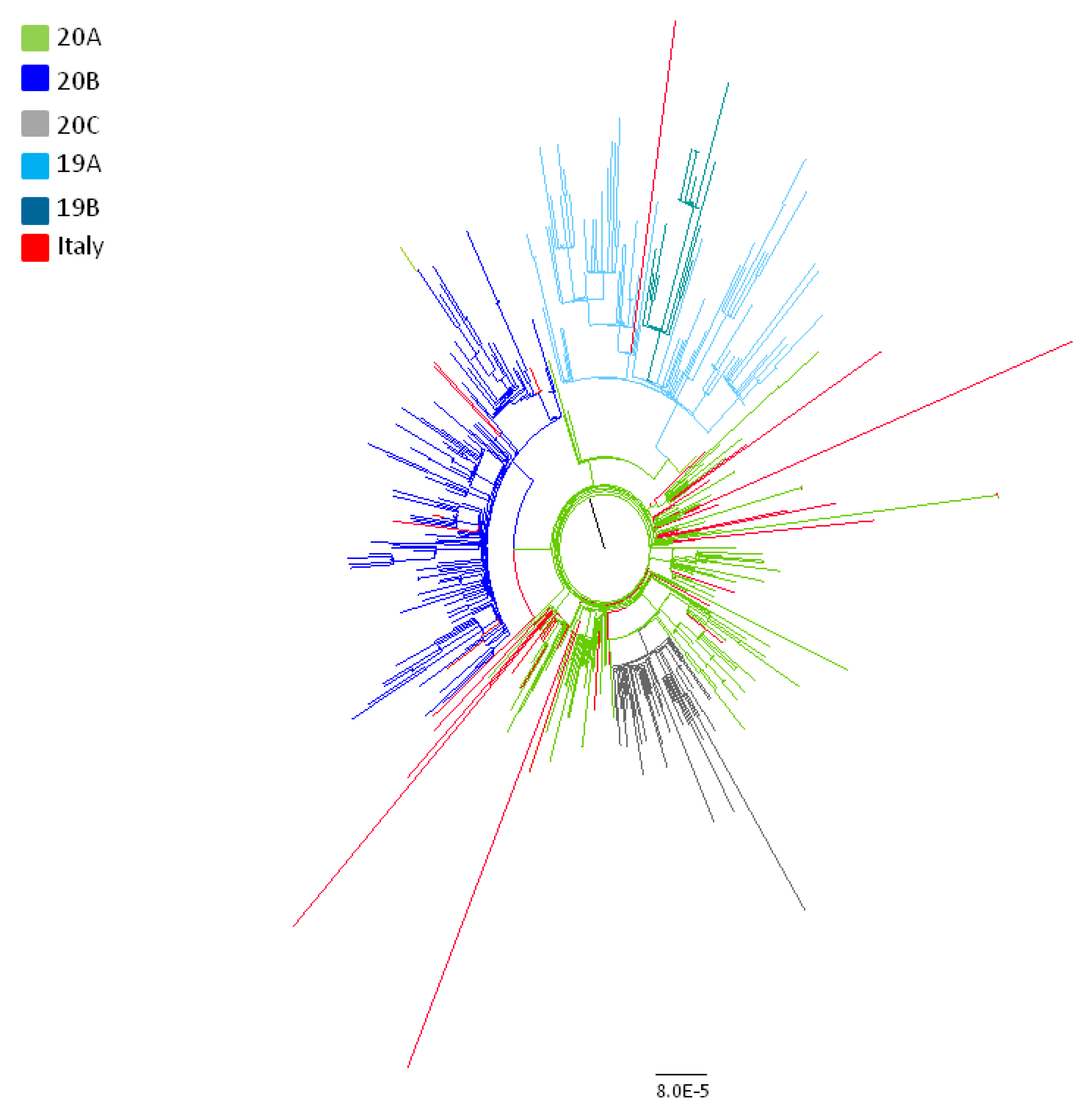
3.1. Phylogenetic Analysis of the Whole Dataset
3.2. Genetic Distances Analysis
3.3. Differences in Amino Acids
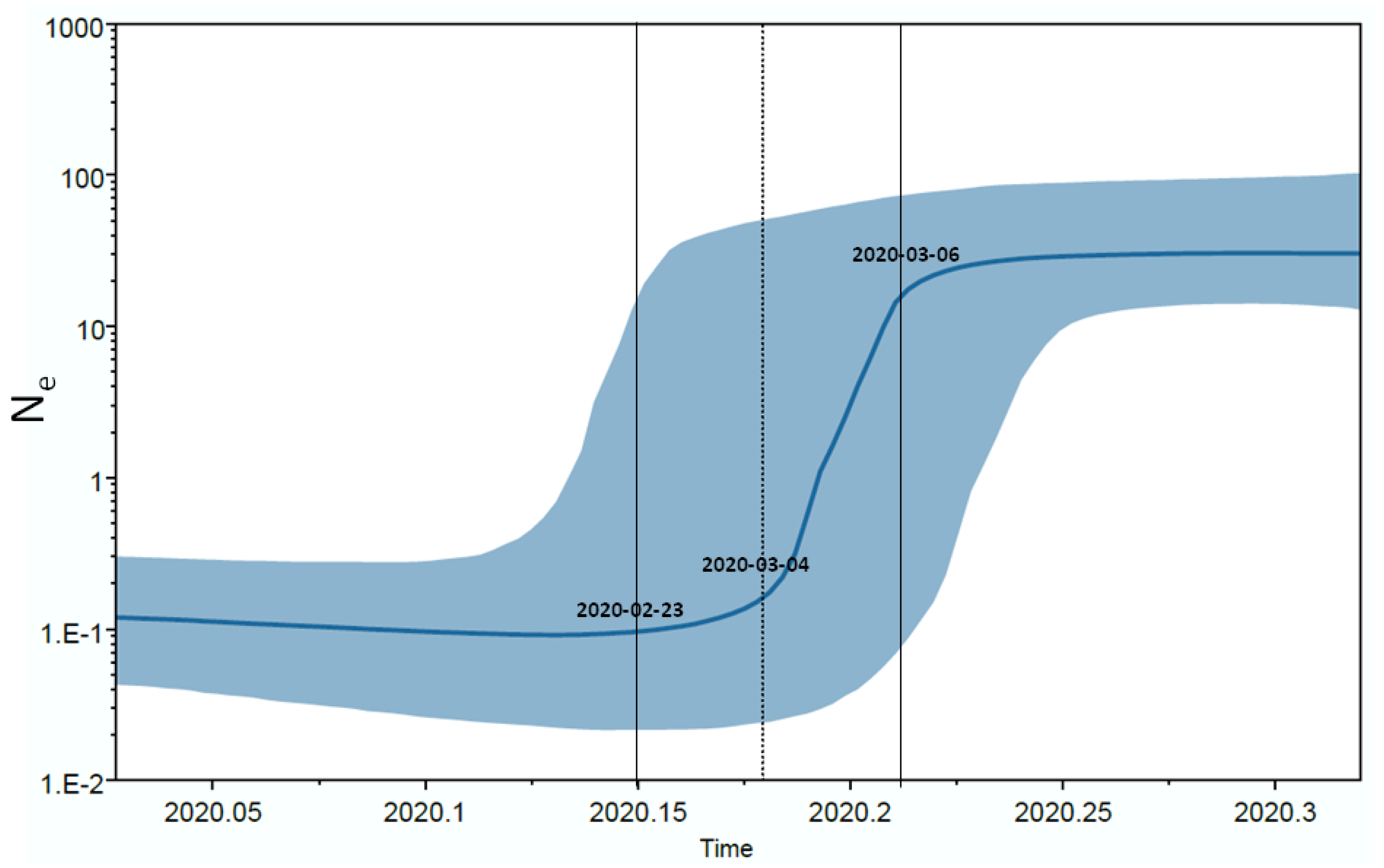
3.4. Time Reconstruction of the SARS-CoV-2 Italian Lineage B.1 Phylogeny
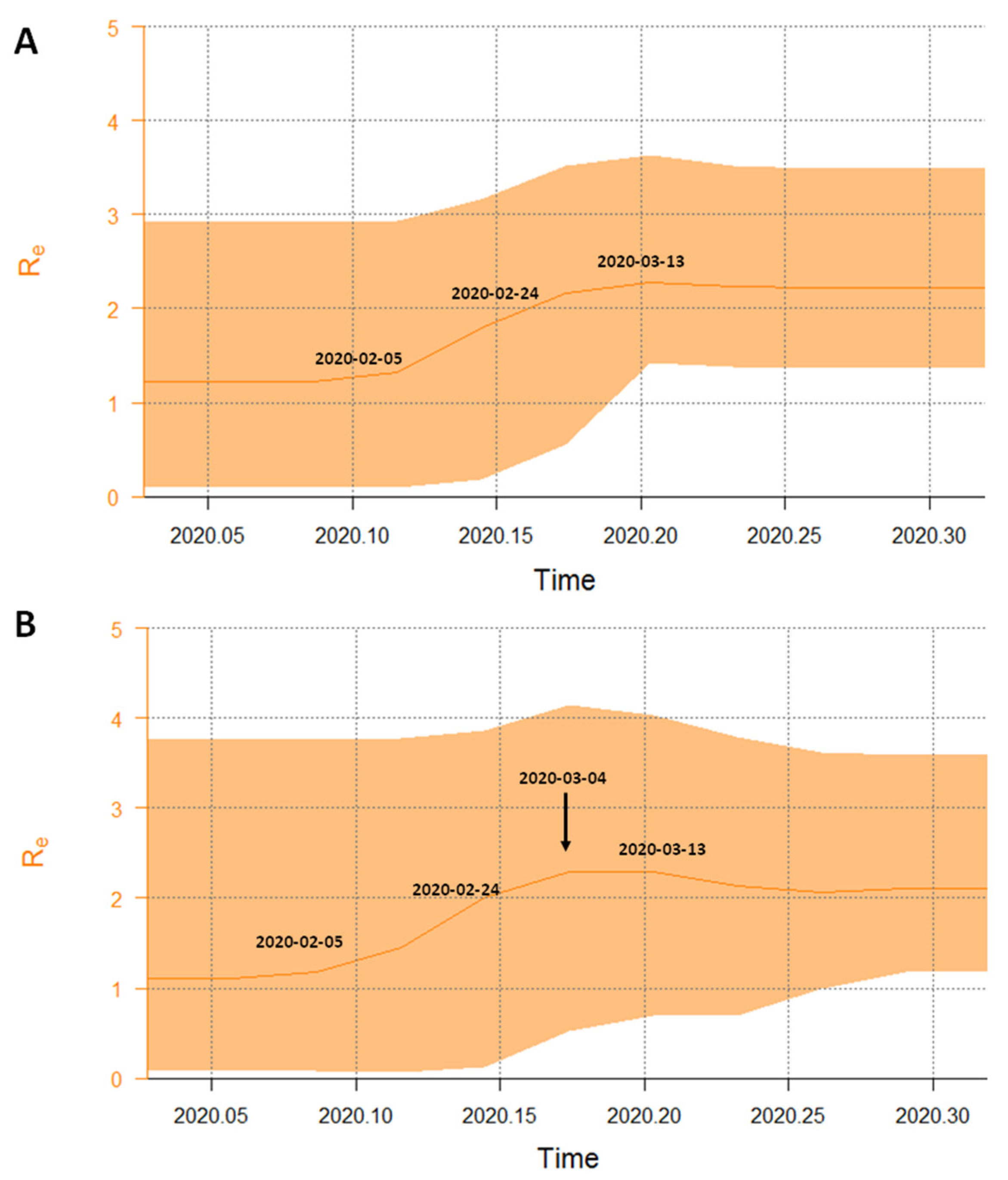
3.5. Phylodynamic Analysis of the Italian Dataset
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Zehender, G.; Lai, A.; Bergna, A.; Meroni, L.; Riva, A.; Balotta, C.; Tarkowski, M.; Gabrieli, A.; Bernacchia, D.; Rusconi, S.; et al. Genomic characterization and phylogenetic analysis of SARS-COV-2 in Italy. J. Med. Virol. 2020, 29, 25794. [Google Scholar] [CrossRef] [Green Version]
- Capobianchi, M.R.; Rueca, M.; Messina, F.; Giombini, E.; Carletti, F.; Colavita, F.; Castilletti, C.; Lalle, E.; Bordi, L.; Vairo, F.; et al. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin. Microbiol. Infect. 2020, 26, 954–956. [Google Scholar] [CrossRef]
- Stefanelli, P.; Faggioni, G.; Presti, A.L.; Fiore, S.; Marchi, A.; Benedetti, E.; Fabiani, C.; Anselmo, A.; Ciammaruconi, A.; Fortunato, A.; et al. Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: Additional clues on multiple introductions and further circulation in Europe. Eurosurveillance 2020, 25, 2000305. [Google Scholar] [CrossRef] [Green Version]
- Rothe, C.; Schunk, M.; Sothmann, P.; Bretzel, G.; Froeschl, G.; Wallrauch, C.; Zimmer, T.; Thiel, V.; Janke, C.; Guggemos, W.; et al. Transmission of 2019-nCoV Infection from an Asymptomatic Contact in Germany. N. Engl. J. Med. 2020, 382, 970–971. [Google Scholar] [CrossRef] [Green Version]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Boil. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Lott, M.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Boil. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.; Hill, V.; O’Toole, Á.; McCrone, J.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv 2020. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Boil. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Boil. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Weiss, R.E.; Sinsheimer, J.S. Bayesian selection of continuous-time Markov chain evolutionary models. Mol. Boil. Evol. 2001, 18, 1001–1013. [Google Scholar] [CrossRef] [Green Version]
- Stadler, T.; Kühnert, D.; Bonhoeffer, S.; Drummond, A.J. Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc. Natl. Acad. Sci. USA 2012, 110, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.; Lau, E.H.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Walker, P.; Pybus, O.G.; Rambaut, A.; Holmes, E. Comparative population dynamics of HIV-1 subtypes B and C: Subtype-specific differences in patterns of epidemic growth. Infect. Genet. Evol. 2005, 5, 199–208. [Google Scholar] [CrossRef]
- Spiteri, G.; Fielding, J.; Diercke, M.; Campese, C.; Enouf, V.; Gaymard, A.; Bella, A.; Sognamiglio, P.; Moros, M.J.S.; Riutort, A.N.; et al. First cases of coronavirus disease 2019 (COVID-19) in the WHO European Region, 24 January to 21 February 2020. Eurosurveillance 2020, 25, 2000178. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; A Gayle, A.; Wilder-Smith, A.; Rocklöv, J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J. Travel Med. 2020, 27, 27. [Google Scholar] [CrossRef] [Green Version]
- D’Arienzo, M.; Coniglio, A. Assessment of the SARS-CoV-2 basic reproduction number, R0, based on the early phase of COVID-19 outbreak in Italy. Biosaf. Heal. 2020. [Google Scholar] [CrossRef]
- Gatto, M.; Bertuzzo, E.; Mari, L.; Miccoli, S.; Carraro, L.; Casagrandi, R.; Rinaldo, A. Spread and dynamics of the COVID-19 epidemic in Italy: Effects of emergency containment measures. Proc. Natl. Acad. Sci. USA 2020, 117, 10484–10491. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Li, M.; Lv, G.; Lu, Z.K. Monitoring transmissibility and mortality of COVID-19 in Europe. Int. J. Infect. Dis. 2020, 95, 311–315. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Acciarri, C.; Galli, M.; Zehender, G. Early phylogenetic estimate of the effective reproduction number of SARS-CoV-2. J. Med. Virol. 2020, 92, 675–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage (Pangolin) | Total | % | From | Nextstrain New | Nextstrain Old |
|---|---|---|---|---|---|
| B | 1 | 1.6 | PD (1) | 19A | nd |
| B.1 | 47 | 75.8 | MI (15), PS(7), AN (1), MC (1) PD (8), BG (1), CR (3), SI (3), AR (3), GR (1), BS (4) | 20A, nd | A2a |
| B.1.1 | 11 | 17.7 | MI (4), PD (1), SI (4), GR (1), AR (1) | 20B | A2a |
| B.1.34 | 1 | 1.6 | MI (1) | nd | A2a |
| B.1.5 | 2 | 3.2 | MI (1), BG (1) | 20A | A2a |
| Time | Within | Time | Between | ||||||
|---|---|---|---|---|---|---|---|---|---|
| p Distance (SE) | Nucleotide (SE) | dS (SE) | dN (SE) | p Distance (SE) | Nucleotide (SE) | dS (SE) | dN (SE) | ||
| February | 3.8 | 9.6 | 3.5 | 3.8 | February vs. March | 3.1 | 8.1 | 2.9 | 2.8 |
| (0.6) | (1.5) | (1.1) | (0.6) | (0.4) | (1.3) | (0.8) | (0.4) | ||
| March | 1.9 | 5.4 | 2.2 | 1.5 | March vs. April | 2.3 | 6.6 | 2.1 | 2.0 |
| (0.3) | (0.8) | (0.5) | (0.4) | (0.3) | (0.8) | (0.6) | (0.5) | ||
| April | 2.4 | 6.8 | 1.7 | 2.1 | February vs. April | 3.7 | 10 | 2.7 | 3.5 |
| (0.3) | (0.9) | (0.8) | (0.5) | (0.5) | (1.5) | (0.8) | (0.6) | ||
| Genome Region | Mutation | n/Total | Percentage (%) |
|---|---|---|---|
| ORF 1ab | S443F | 2/135 | 1.5 |
| H3076Y | 2/135 | 1.5 | |
| L3606F | 3/131 | 2.3 | |
| P4715L | 133/136 | 97.8 | |
| E5689D | 2/135 | 1.5 | |
| R5919K | 2/123 | 1.6 | |
| S | A570D | 2/129 | 1.6 |
| D614G | 128/130 | 98.5 | |
| G1046V * | 3/134 | 2.2 | |
| ORF 3a | G251V | 3/134 | 2.2 |
| M | D3G | 21/133 | 15.8 |
| ORF 7a | G70C | 2/134 | 1.5 |
| N | R203K-G204R | 52/133 | 39.1 |
| V246I | 3/136 | 2.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, A.; Bergna, A.; Caucci, S.; Clementi, N.; Vicenti, I.; Dragoni, F.; Cattelan, A.M.; Menzo, S.; Pan, A.; Callegaro, A.; et al. Molecular Tracing of SARS-CoV-2 in Italy in the First Three Months of the Epidemic. Viruses 2020, 12, 798. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080798
Lai A, Bergna A, Caucci S, Clementi N, Vicenti I, Dragoni F, Cattelan AM, Menzo S, Pan A, Callegaro A, et al. Molecular Tracing of SARS-CoV-2 in Italy in the First Three Months of the Epidemic. Viruses. 2020; 12(8):798. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080798
Chicago/Turabian StyleLai, Alessia, Annalisa Bergna, Sara Caucci, Nicola Clementi, Ilaria Vicenti, Filippo Dragoni, Anna Maria Cattelan, Stefano Menzo, Angelo Pan, Annapaola Callegaro, and et al. 2020. "Molecular Tracing of SARS-CoV-2 in Italy in the First Three Months of the Epidemic" Viruses 12, no. 8: 798. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080798