
Signaling Circuits and Regulation of Immune Suppression by Ovarian Tumor-Associated Macrophages

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immune Suppression by Ovarian Tumor-Associated Macrophages
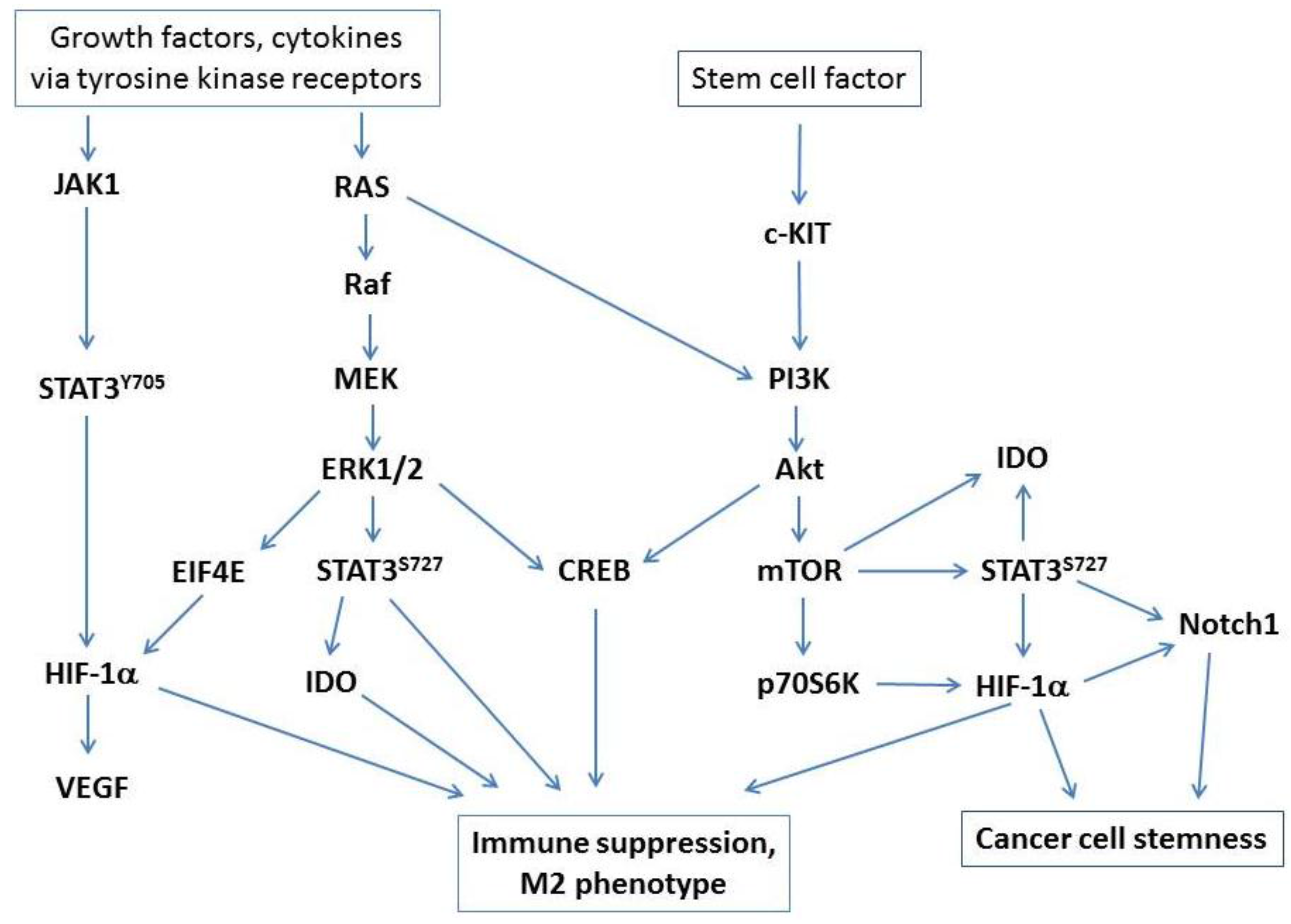
2.1. A Central Role for STAT3
2.2. Hypoxia, Immune Suppression and Tumor Progression

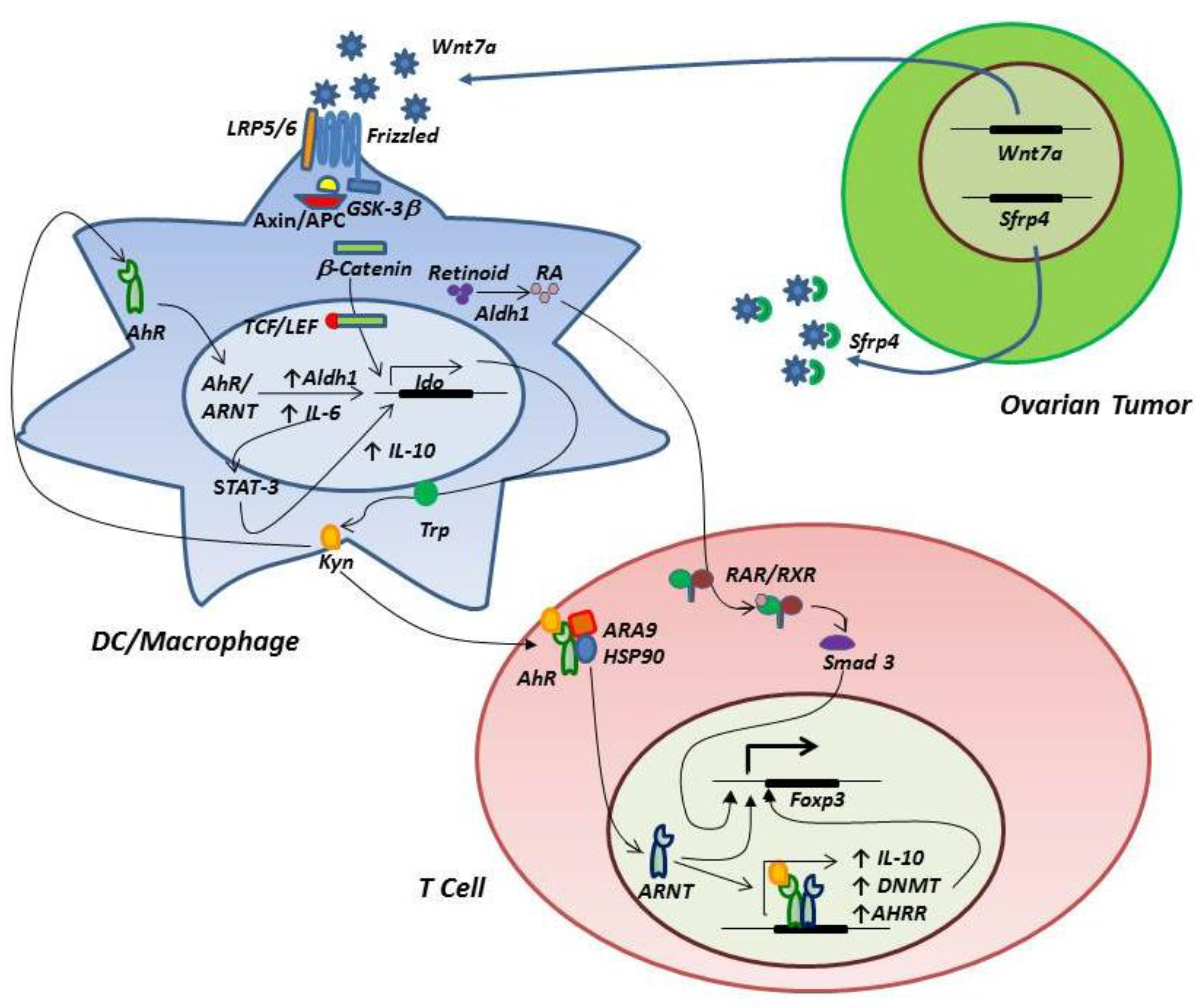
3. Regulation of IDO Expression and Treg Differentiation
3.1. Stem Cell Factor and c-KIT Signaling
3.2. The IDO-AhR Relationship

3.3. Signaling through the Wnt/β-Catenin Pathway
4. Regulation of Co-Inhibitory Molecule Expression
5. Potential Therapeutic Interventions that Target Ovarian Tumor-Associated Macrophage Signaling and IDO Expression
5.1. Inhibitors of c-KIT/PI3K/Akt/mTOR
5.2. Inhibition of IL-6 and STAT3 Signaling
5.3. Inhibition of Wnt/β-Catenin and p38 Signaling
5.4. Direct Inhibition of IDO Function
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Wolf, A.M.; Rumpold, H.; Fiegl, H.; Zeimet, A.G.; Muller-Holzner, E.; Deibl, M.; Gastl, G.; Gunsilius, E.; Marth, C.; et al. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin. Cancer Res. 2005, 11, 8326–8331. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [PubMed]
- Inaba, T.; Ino, K.; Kajiyama, H.; Yamamoto, E.; Shibata, K.; Nawa, A.; Nagasaka, T.; Akimoto, H.; Takikawa, O.; Kikkawa, F.; et al. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol. Oncol. 2009, 115, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Goyne, H.E.; Stone, P.J.B.; Burnett, A.F.; Cannon, M.J. Ovarian tumor ascites CD14+ cells suppress dendritic cell-activated CD4+ T cell responses through IL-10 secretion and indoleamine 2,3-dioxygenase. J. Immunother. 2014, 37, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.M.; Kryczek, I.; Zou, W. Antigen-presenting cell (APC) subsets in ovarian cancer. Int. Rev. Immunol. 2011, 30, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Krempski, J.; Karyampudi, L.; Behrens, M.D.; Erskine, C.L.; Hartmann, L.; Dong, H.; Goode, E.L.; Kalli, K.R.; Knutson, K.L. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J. Immunol. 2011, 186, 6905–6913. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, U.K.; Rutkowski, M.R.; Rauwerdink, A.M.; Fields, J.; Escovar-Fadul, X.; Baird, J.; Cubillos-Ruiz, J.R.; Jacobs, A.C.; Gonzalez, J.L.; Weaver, J.; et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 2012, 209, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Reinartz, S.; Schumann, T.; Finkernagel, F.; Wortmann, A.; Jansen, J.M.; Meissner, W.; Krause, M.; Schwörer, A.-M.; Wagner, U.; Müller-Brüsselbach, S.; et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: Correlation of CD163 expression, cytokine levels and early relapse. Int. J. Cancer 2014, 134, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Wang, H.W.; Cuenca, A.; Huang, M.; Ghansah, T.; Brayer, J.; Kerr, W.G.; Takeda, K.; Akira, S.; Schoenberger, S.P.; et al. A critical role for Stat3 signaling in immune tolerance. Immunity 2003, 19, 425–436. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mule, J.; Kerr, W.G.; et al. Inhibiting STAT3 signaling in the hematopoeitc system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, K.; Komohara, Y.; Tashiro, H.; Ohtake, H.; Nakagawa, T.; Katabuchi, H.; Takeya, M. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010, 101, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Montminy, M. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 1998, 273, 32377–32379. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, D.; Mourkioti, F.; Gambardella, A.; Kirstetter, P.; Lopez, R.G.; Rosenthal, N.; Nerlov, C. A CREB-C/EBPβ cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA 2009, 106, 17475–17480. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S.; et al. Macrophage expression of hypoxia-inducible factor-1α suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Laoui, L.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M.; et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibitis angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Osinsky, S.; Bubnovskaya, L.; Ganusevich, I.; Kovelskaya, A.; Gumenyuk, L.; Olijnichenko, G.; Merentsev, S. Hypoxia, tumour-associated macrophages, microvessel density, VEGF and matrix metalloproteinases in human gastric cancer: Interaction and impact on survival. Clin. Transl. Oncol. 2011, 13, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Selvendiran, K.; Kuppusamy, M.L.; Ahmed, S.; Bratasz, A.; Meenakshisundaram, G.; Rivera, B.K.; Khan, M.; Kuppusamy, P. Oxygenation inhibits ovarian tumor growth by downregulating STAT3 and cyclin-D1 expressions. Cancer Biol. Ther. 2010, 10, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.S.; Munkarah, A.R.; Kumar, S.; Batchu, R.B.; Shah, J.P.; Berman, J.; Morris, R.T.; Jiang, Z.L.; Saed, G.M. Reduction of hypoxia-induced angiogenesis in ovarian cancer cells by inhibition of HIH-1 alpha gene expression. Arch. Gynecol. Obstet. 2010, 282, 677–683. [Google Scholar] [PubMed]
- Gustaffson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M.; et al. Hypoxia requires Notch signaling to maintain the undifferentiated state. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wu, T.; Zhang, H.-W.; Lu, N.; Wang, Y.-J.; Zhao, L.; Chen, F.-H.; Wang, X.-T.; You, Q.-D.; Guo, Q.-L.; et al. HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Ma, Y.; Liu, J.; Trope, C.G.; Holm, R.; Nesland, J.M.; Suo, Z. The hypoxic microenvironment upgrades stem-like properties of ovarian cancer cells. BMC Cancer 2012. [Google Scholar] [CrossRef] [PubMed]
- Panni, R.Z.; Sanford, D.E.; Belt, B.A.; Mitchem, J.B.; Worley, L.A.; Goetz, B.D.; Mukherjee, P.; Wang-Gillam, A.; Link, D.C.; Denardo, D.G.; et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol. Immunother. 2014, 63, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Pawlus, M.R.; Wang, L.; Hu, C.J. STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.D.; Hou, D.Y.; Liu, Y.; Koni, P.A.; Metz, R.; Chandler, P.; Mellor, A.L.; He, Y.; Munn, D.H. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 2009, 113, 6102–6111. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.J.; Rossi, M.; Romano, E.; Ghith, J.; Yuan, J.; Munn, D.H.; Young, J.W. Indoleamine 2,3-dioxygenase-expressing mature human monocyte-derived dendritic cells expand potent autologous regulatory T cells. Blood 2009, 114, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Raiyan, C.; Rossi, F.; et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Kyo, S.; Fujita, M.; Enomoto, T.; Kondoh, G. Coexpression of the c-kit receptor and the stem cell factor in gynecological tumors. Cancer Res. 1994, 54, 3049–3053. [Google Scholar] [PubMed]
- Parrott, J.A.; Kim, G.; Skinner, M.K. Expression and action of kit ligand/stem cell factor in normal human and bovine ovarian surface epithelium and ovarian cancer. Biol. Reprod. 2000, 62, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Wilczynski, S.P.; Chen, Y.-Y.; Chen, W.; Howell, S.B.; Shively, J.E.; Alberts, D.S. Expression and mutational analysis of tyrosine kinase receptors c-kit, PDGFRα, and PDGFRβ in ovarian cancers. Hum. Pathol. 2005, 36, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Goyne, H.E.; Cannon, M.J. Dendritic cell vaccination, immune regulation and clinical outcomes in ovarian cancer. Front. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Funatake, C.J.; Marshall, N.B.; Steppan, L.B.; Mourich, D.V.; Kerkvliet, N.I. Cutting edge: Activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+CD25+ cells with characteristics of regulatory T cells. J. Immunol. 2005, 175, 4184–4188. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.; Kumar, D.; Burns, E.J.; Nadeau, M.; Dake, B.; Laroni, A.; Kozoriz, D.; Weiner, H.L.; Quintana, F.J. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3+ regulatory T cells. Nat. Immunol. 2010, 11, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.B.; Kerkvliet, N. Dioxin and immune regulation: Emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann. N. Y. Acad. Sci. 2010, 1183, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.-C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farex, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.P.; Singh, U.P.; Singh, B.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS ONE 2011, 6, e23522. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Murugaiyan, G.; Farez, M.F.; Mitsdoerffer, M.; Tukpah, A.M.; Burns, E.J.; Weiner, H.L. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 107, 20768–20773. [Google Scholar] [CrossRef] [PubMed]
- Pot, C. Aryl hydrocarbon receptor controls regulatory CD4+ T cell function. Swiss Med. Wkly. 2012. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [PubMed]
- Yoshioka, S.; King, M.L.; Ran, S.; Okuda, H.; MacLean, J.A., 2nd; McAsey, M.E.; Sugino, N.; Brard, L.; Watabe, K.; Hayashi, K.; et al. WNT7A regulates tumor growth and progression of ovarian cancer through the WNT/β-catenin pathway. Mol. Cancer Res. 2012, 10, 469–482. [Google Scholar] [PubMed]
- Arend, R.C.; Londoño-Joshi, A.I.; Straughn, J.M., Jr.; Buchsbaum, D.J. The wnt/β-catenin pathway in ovarian cancer: A review. Gynecol. Oncol. 2013, 131, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Peng, C.J.; Peng, J.; Jiang, L.Y.; Ning, X.M.; Zheng, J.H. Prognostic role of Wnt7a expression in ovarian carcinoma patients. Neoplasma 2010, 57, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.-J.; Cassetta, L.; Qian, B.-Z.; Lewkowich, I.; Li, J.-F.; Stefater, J.A., 3rd; Smith, A.N.; Wiechmann, L.S.; Wang, Y.; Pollard, J.W.; et al. Myeloid WNT7b mediates the angiogenic switch and metastasis in breast cancer. Cancer Res. 2014, 74, 2962–2973. [Google Scholar] [CrossRef] [PubMed]
- Schaale, K.; Neumann, J.; Schneider, D.; Ehlers, S.; Reiling, N. Wnt signaling in macrophages: Augmenting and inhibiting mycobacteria-induced inflammatory responses. Eur. J. Cell Biol. 2011, 90, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Manicassamy, S.; Reizis, B.; Ravindran, R.; Nakaya, H.; Salazar-Gonzalez, R.M.; Wang, Y.C.; Pulendran, B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 2011, 329, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Ukegjini, K.; Nixdorf, S.; Ford, C.E.; Olivier, J.; Caduff, R.; Scurry, J.P.; Guertler, R.; Hornung, D.; Muller, R.; et al. Loss of secreted frizzled-related protein 4 correlates with an aggressive phenotype and predicts poor outcome in ovarian cancer patients. PLoS ONE 2012, 7, e31885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saran, U.; Arfuso, F.; Zeps, N.; Dharmarajan, A. Secreted frizzled-related protein 4 expression is positively associated with responsiveness to cisplatin of ovarian cancer cell lines in vitro and with lower tumour grade in mucinous ovarian cancers. BMC Cell Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Thornton, T.M.; Pedraza-Alva, G.; Deng, B.; Wood, C.D.; Aronshtam, A.; Clements, J.L.; Sabio, G.; Davis, R.J.; Matthews, D.E.; Doble, B.; et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3β inactivation. Science 2008, 320, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Bikkavilli, R.K.; Feigin, M.E.; Malbon, C.C. p38 Mitogen-activated protein kinase regulates canonical Wnt-b-catenin signaling by inactivation of GSK3β. J. Cell Sci. 2008, 121, 3598–3607. [Google Scholar] [CrossRef] [PubMed]
- Soichot, M.; Hennart, B.; al Saabi, A.; Leloire, A.; Froguel, P.; Levy-Marchal, C.; Poulain-Godefroy, O.; Allorge, D. Identification of a variable number of tandem repeats polymorphism and characterization of LEF-1 response elements in the promoter of the IDO1 gene. PLoS ONE 2011, 6, e25470. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.J.; Goyne, H.E.; Stone, P.J.B.; MacDonald, L.J.; James, L.E.; Cobos, E.; Chiriva-Internati, M. Modulation of p38 MAPK signaling enhances dendritic cell activation of human CD4+ Th17 responses to ovarian tumor antigen. Cancer Immunol. Immunother. 2013, 62, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with Lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus Ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Wolfle, S.J.; Strebovsky, J.; Bartz, H.; Sahr, A.; Arnold, C.; Kaiser, C.; Dalpke, A.H.; Heeg, K. PD-L1 expression on tolerogenic APCs is controlled by STAT3. Eur. J. Immunol. 2011, 41, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Zou, L.; Rodriguez, P.; Zhu, G.; Wei, S.; Mottram, P.; Brumlik, M.; Cheng, P.; Curiel, T.; Myers, L.; et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J. Exp. Med. 2006, 203, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wei, S.; Zhu, G.; Myers, L.; Mottram, P.; Cheng, P.; Chen, L.; Coukos, G.; Zou, W. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res. 2007, 67, 8900–8905. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wei, S.; Zou, L.; Zhu, G.; Mottram, P.; Xu, H.; Chen, L.; Zou, W. Cutting edge: Induction of B7-H4 on APCs through IL-10: Novel suppressive mode for regulatory T cells. J. Immunol. 2006, 177, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cheng, F.; Cuenca, A.; Horna, P.; Zheng, Z.; Bhalla, K.; Sotomayor, E.M. Imatinib mesylate (STI-571) enhances antigen-presenting cell function and overcomes tumor-induced CD4+ T-cell tolerance. Blood 2005, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Larmonier, N.; Janikashvili, N.; LaCasse, C.J.; Larmonier, C.B.; Cantrell, J.; Situ, E.; Lundeen, T.; Bonnotte, B.; Katsanis, E. Imatinib mesylate inhibits CD4+CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL− tumors. J. Immunol. 2008, 181, 6955–6963. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.; Vanderhyden, B.C. AKT mediates the pro-survival effects of KIT in ovarian cancer cells and is a determinant of sensitivity to imatinib mesylate. Gynecol. Oncol. 2007, 105, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Broaddus, R.R.; Bodurka, D.C.; Wolf, J.K.; Burke, T.W.; Kavanagh, J.J.; Levenback, C.F.; Gershenson, D.M. Phase II trial of imatinib mesylate in patients with recurrent platimun- and taxane-resistant epithelial ovarian and primary peritoneal cancers. Gynecol. Oncol. 2006, 101, 126–131. [Google Scholar] [PubMed]
- Schilder, R.J.; Sill, M.W.; Lee, R.B.; Shaw, T.J.; Senterman, M.K.; Klein-Szanto, A.J.; Miner, Z.; Vanderhyden, B.C. Phase II evaluation of imatinib mesylate in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2008, 26, 3418–3425. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Adams, S. Small-Molecule protein kinase inhibitors and their effects on the immune system: Implications for cancer treatment. Immunotherapy 2011, 3, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; George, J.; Kulbe, H.; Friedlander, M.; Rischin, D.; Lemech, C.; Power, J.; Coward, J.; Cowin, P.A.; House, C.M.; et al. IL6-STAT3-HIF signaling and therapeutic response to the angiogenesis inhibitor sunitinib in ovarian clear cell cancer. Clin. Cancer Res. 2011, 17, 2538–2548. [Google Scholar] [CrossRef] [PubMed]
- Biagi, J.J.; Oza, A.M.; Chachal, H.I.; Grimshaw, R.; Ellard, S.L.; Lee, U.; Hirte, H.; Sederias, J.; Ivy, S.P.; Eisenhauer, E.A.; et al. A phase II study of sunitinib in patients with recurrent epithelial ovarian and primary peritoneal carcinoma: An NCIC Clinical Trials Group Study. Ann. Oncol. 2011, 22, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Zhang, C.; Herrmann, A.; Du, Y.; Figlin, R.; Yu, H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009, 69, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Ozao-Choy, J.; Ma, G.; Kao, J.; Wang, G.X.; Meseck, M.; Sung, M.; Schwartz, M.; Divino, C.M.; Pan, P.-Y.; Chen, S.-H.; et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009, 69, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Finke, J.H.; Rini, B.; Ireland, J.; Rayman, P.; Richmond, A.; Golshayan, A.; Wood, L.; Elson, P.; Garcia, J.; Dreicer, R.; et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin. Cancer Res. 2008, 14, 6674–6682. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.S.; Zea, A.H.; Rini, B.R.; Ireland, J.I.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib mediates reversal of myeloid-derived suppressor cells accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.-Y.; Subjeck, J.R.; Shikant, P.; Kim, H.L. Temsirolimus, an mTOR inhibitor, enhances anti-tumour effects of heat shock protein cancer vaccines. Br. J. Cancer 2011, 104, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Land, S.C.; Tee, A.R. Hypoxia-indicible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed]
- Coward, J.; Kulbe, H.; Chakravarty, P.; Leader, D.; Vassileva, V.; Leinster, D.A.; Thompson, R.; Schioppa, T.; Nemeth, J.; Vermeulen, J.; et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin. Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Hong, D.S.; Burris, H.A., 3rd; Naing, A.; Jones, S.F.; Falchook, G.; Bricmont, P.; Elekes, A.; Rock, E.P.; Kurzrock, R. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 125–130. [Google Scholar] [PubMed]
- Okusaka, T.; Ueno, H.; Ikeda, M.; Mitsunaga, S.; Ozaka, M.; Ishii, H.; Yokosuka, O.; Ooka, Y.; Yoshimoto, R.; Yanagihara, Y.; et al. Phase I and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor, in patients with advanced hepatocellular carcinoma. Hepatol. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jougasaki, M.; Ichiki, T.; Takenoshita, Y.; Setoguchi, M. Statins suppress interleukin-6-induced monocyte chemo-attractant protein-1 by inhibiting Janus kinase/signal transducers and activators of transcription pathways in human vascular endothelial cells. Br. J. Pharmacol. 2010, 159, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Loppnow, H.; Zhang, L.; Buerke, M.; Lautenschlager, M.; Chen, L.; Frister, A.; Schlitt, A.; Luther, T.; Song, N.; Hofmann, B.; et al. Statins potently reduce the cytokine-mediated IL-6 release in SMC/MNC cocultures. J. Cell. Mol. Med. 2011, 15, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Habis, M.; Wroblewski, K.; Bradaric, M.; Ismail, N.; Yamada, S.D.; Litchfield, L.; Lengyel, E.; Romero, I.L. Statin therapy is associated with improved survival in patients with non-serous-papillary epithelial ovarian cancer: A retrospective cohort analysis. PLoS ONE 2014, 9, e104521. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.L.; Kim, H.Y.; Moon, S.H.; et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.M.; Anderson, B.D.; Brooks, N.A.; Brooks, H.B.; Chan, E.M.; de Dios, A.; Gilmour, R.; Graff, J.R.; Jambrina, E.; Mader, M.; et al. Characterization of LY2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity. Mol. Cancer Ther. 2014, 13, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Tanizaki, Y.; Kobayashi, A.; Toujima, S.; Shiro, M.; Mizoguchi, M.; Mabuchi, Y.; Yagi, S.; Minami, S.; Takikawa, O.; Ino, K.; et al. Indoleamine 2,3-dioxygenase promotes peritoneal metastasis of ovarian cancer by inducing an immunosuppressive environment. Cancer Sci. 2014, 105, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.H.; Jackson, E.; Neuger, T.; Dees, E.C.; Harvey, R.D.; Han, H.; Ismail-Khan, R.; Minton, S.; Vahanian, N.N.; Link, C.; et al. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget 2014, 5, 8136–8146. [Google Scholar] [PubMed]
- Mautino, M.R.; Jaipuri, F.A.; Waldo, J.; Kumar, S.; Adams, J.; van Allen, C.; Marcinowicz-Flick, A.; Munn, D.; Vahanian, N.; Link, C.J.; et al. Abstract 491: NLG919, a novel indoleamine-2,3-dioxygenase (IDO)-pathway inhibitor drug candidate for cancer therapy. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Liu, X.; Shin, N.; Koblish, H.K.; Yang, G.; Wang, Q.; Wang, K.; Leffet, L.; Hansbury, M.J.; Thomas, B.; Rupar, M.; et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010, 115, 3520–3530. [Google Scholar] [CrossRef] [PubMed]
- Koblish, H.K.; Hansbury, M.J.; Bowman, K.J.; Yang, G.; Neilan, C.L.; Haley, P.J.; Burn, T.C.; Waeltz, P.; Sparks, R.B.; Yue, E.W.; et al. Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and the growth of IDO-expressing tumors. Mol. Cancer Ther. 2010, 9, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Banerjee, M.; Cheng, P.; Vatan, L.; Szeliga, W.; Wei, S.; Huang, E.; Finlayson, E.; Simeone, D.; Welling, T.H.; et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 2009, 114, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cannon, M.J.; Ghosh, D.; Gujja, S. Signaling Circuits and Regulation of Immune Suppression by Ovarian Tumor-Associated Macrophages. Vaccines 2015, 3, 448-466. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines3020448
Cannon MJ, Ghosh D, Gujja S. Signaling Circuits and Regulation of Immune Suppression by Ovarian Tumor-Associated Macrophages. Vaccines. 2015; 3(2):448-466. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines3020448
Chicago/Turabian StyleCannon, Martin J., Debopam Ghosh, and Swetha Gujja. 2015. "Signaling Circuits and Regulation of Immune Suppression by Ovarian Tumor-Associated Macrophages" Vaccines 3, no. 2: 448-466. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines3020448