The Design and Development of a Multi-HBV Antigen Encoded in Chimpanzee Adenoviral and Modified Vaccinia Ankara Viral Vectors; A Novel Therapeutic Vaccine Strategy against HBV

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. HBV Immunogens
2.2. Plasmids
2.3. HBV Immunogen Expression Analysis in Western Blots
2.4. Vaccines
2.5. Mutant HBV-Polymerase Functionality Analysis
2.6. Animal Experiments
2.6.1. Mice
2.6.2. Intervention
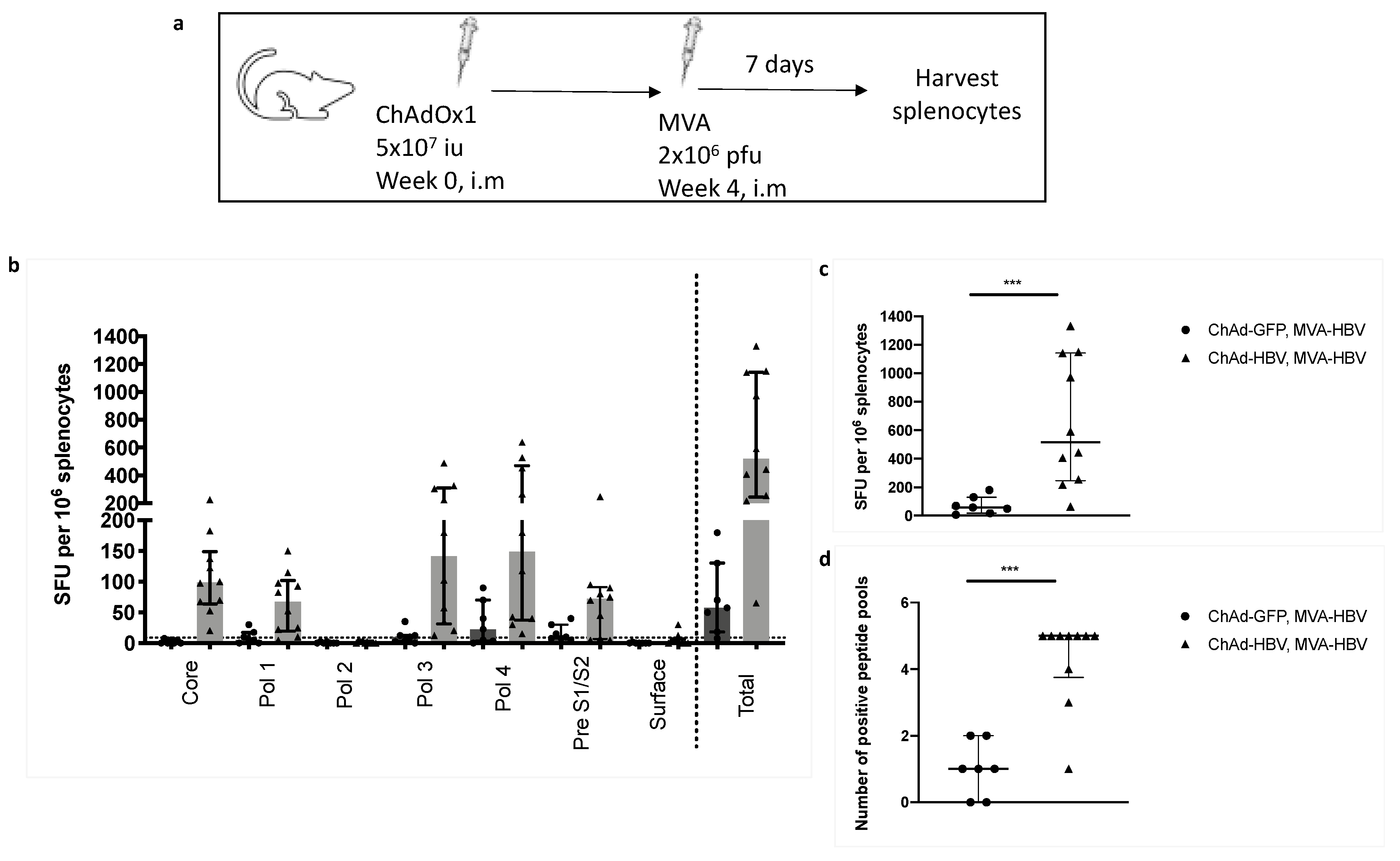
2.6.3. Experimental Design
2.7. Peptides
2.8. Splenocyte and Intra Hepatic Lymphocyte Isolation
2.9. Ex-Vivo IFN ELISpots
2.10. Intracellular Cytokine Staining
2.11. ELISA
2.11.1. Anti-HBs ELISA
2.11.2. PreS1 and HBs Antigen Capture ELISA
2.12. Statistical Analyses
3. Results
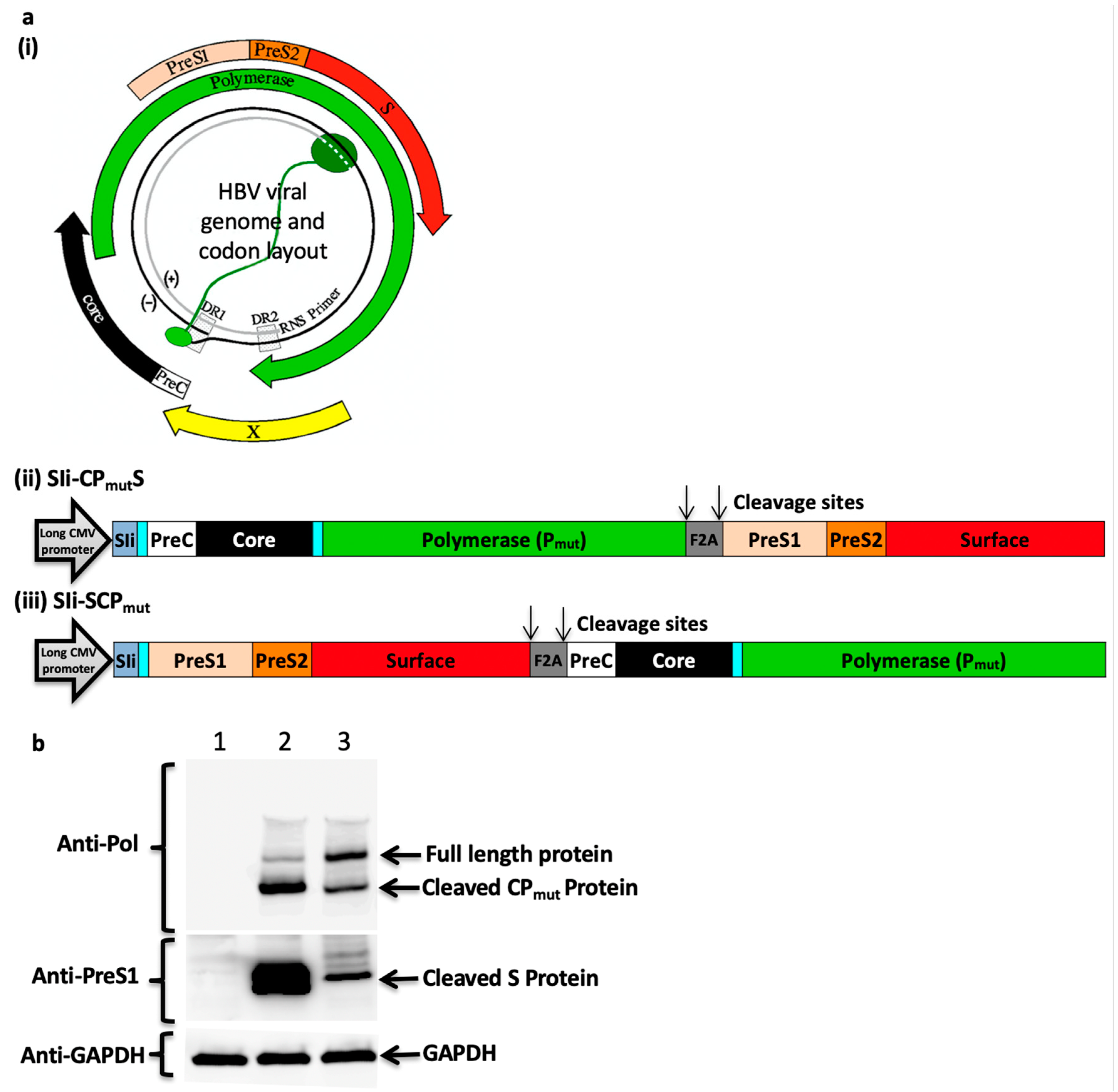
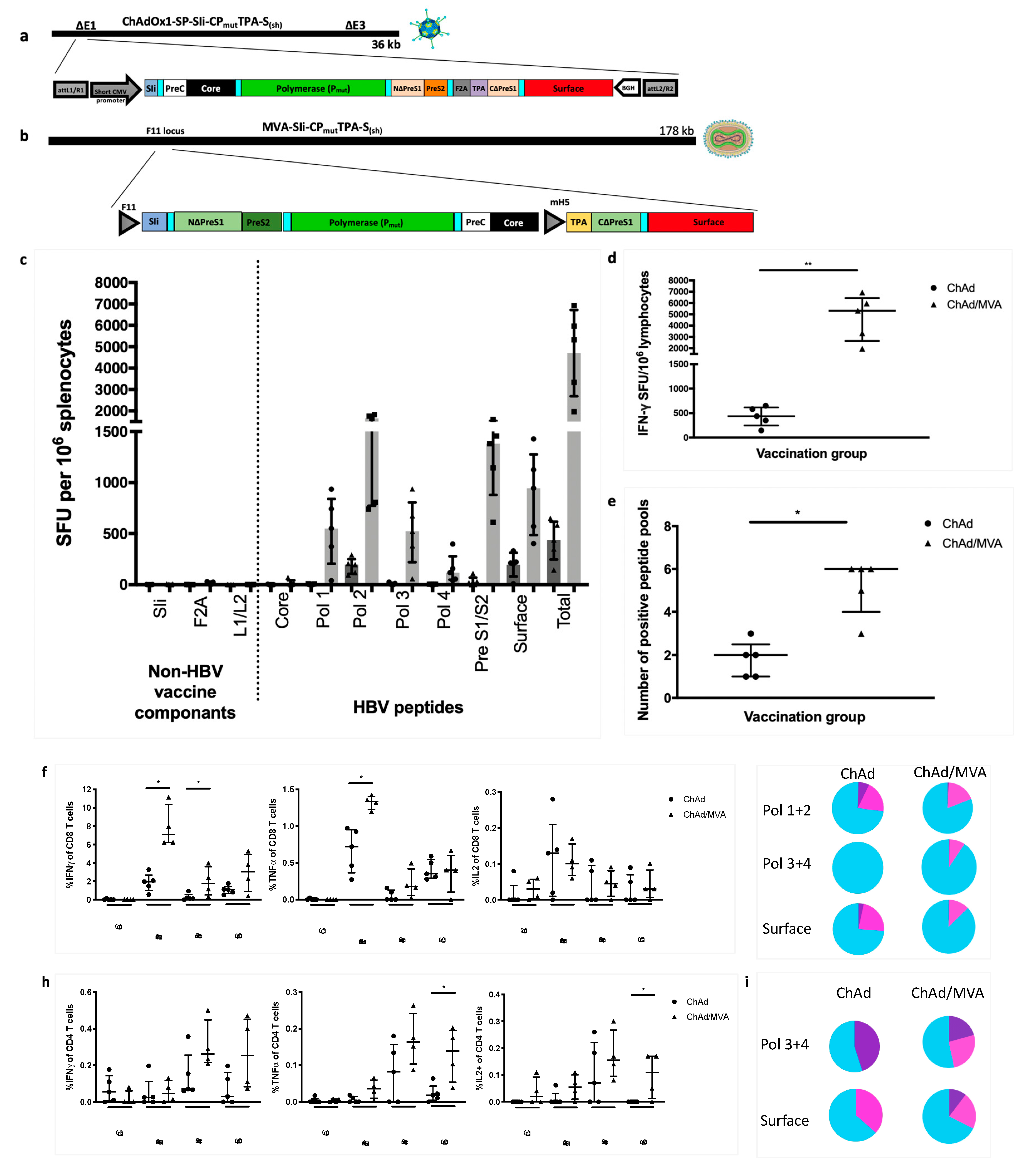
3.1. Generation of the HBV Immunogen
3.2. Furin 2A Inclusion Enables the Generation of Two Separate Polypeptides from a Single Immunogen
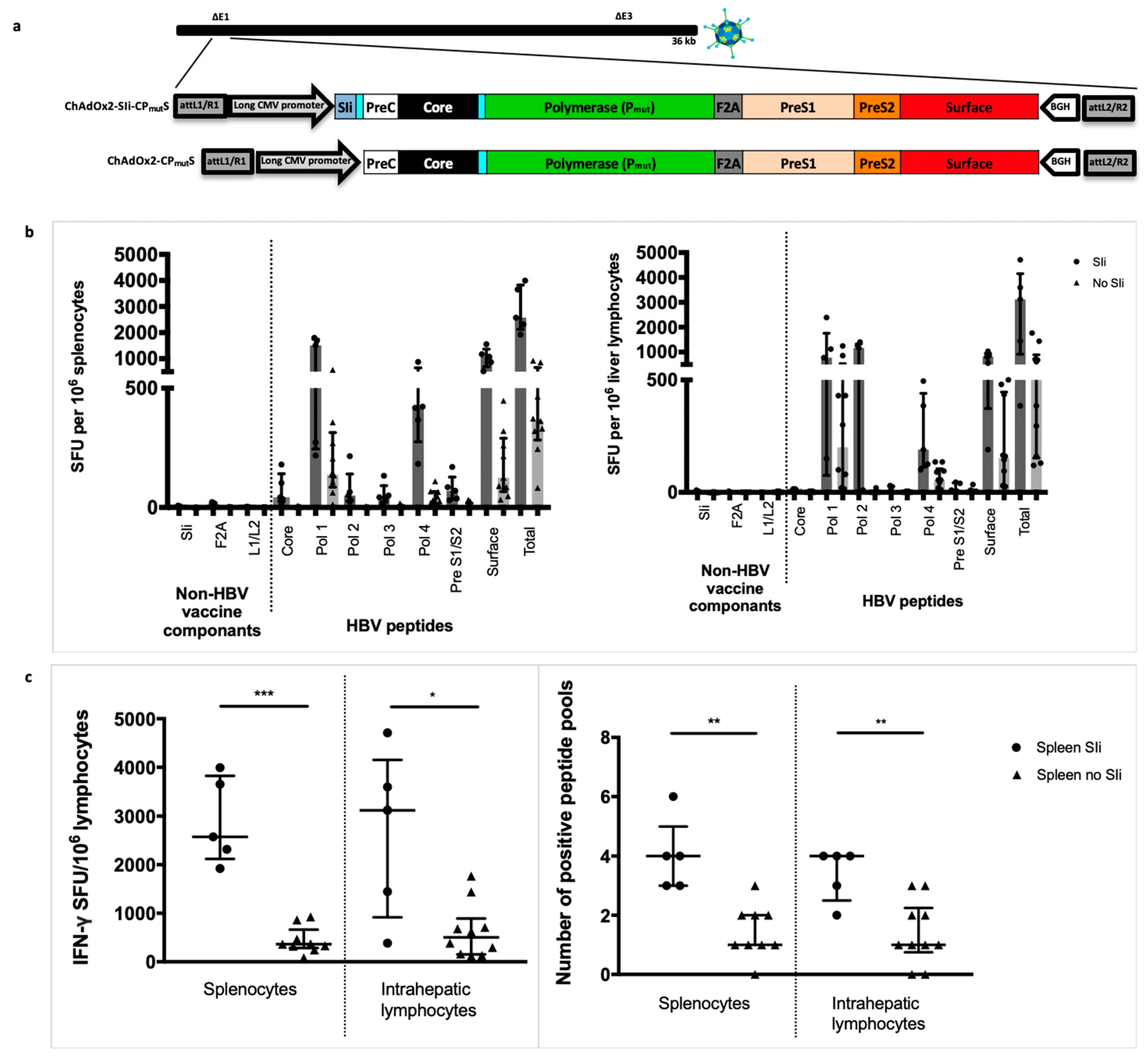
3.3. Addition of the Transmembrane Region of Shark Invariant Chain to the HBV Immunogen Enhances the Magnitude and Breadth of Vaccine Induced T Cell Responses
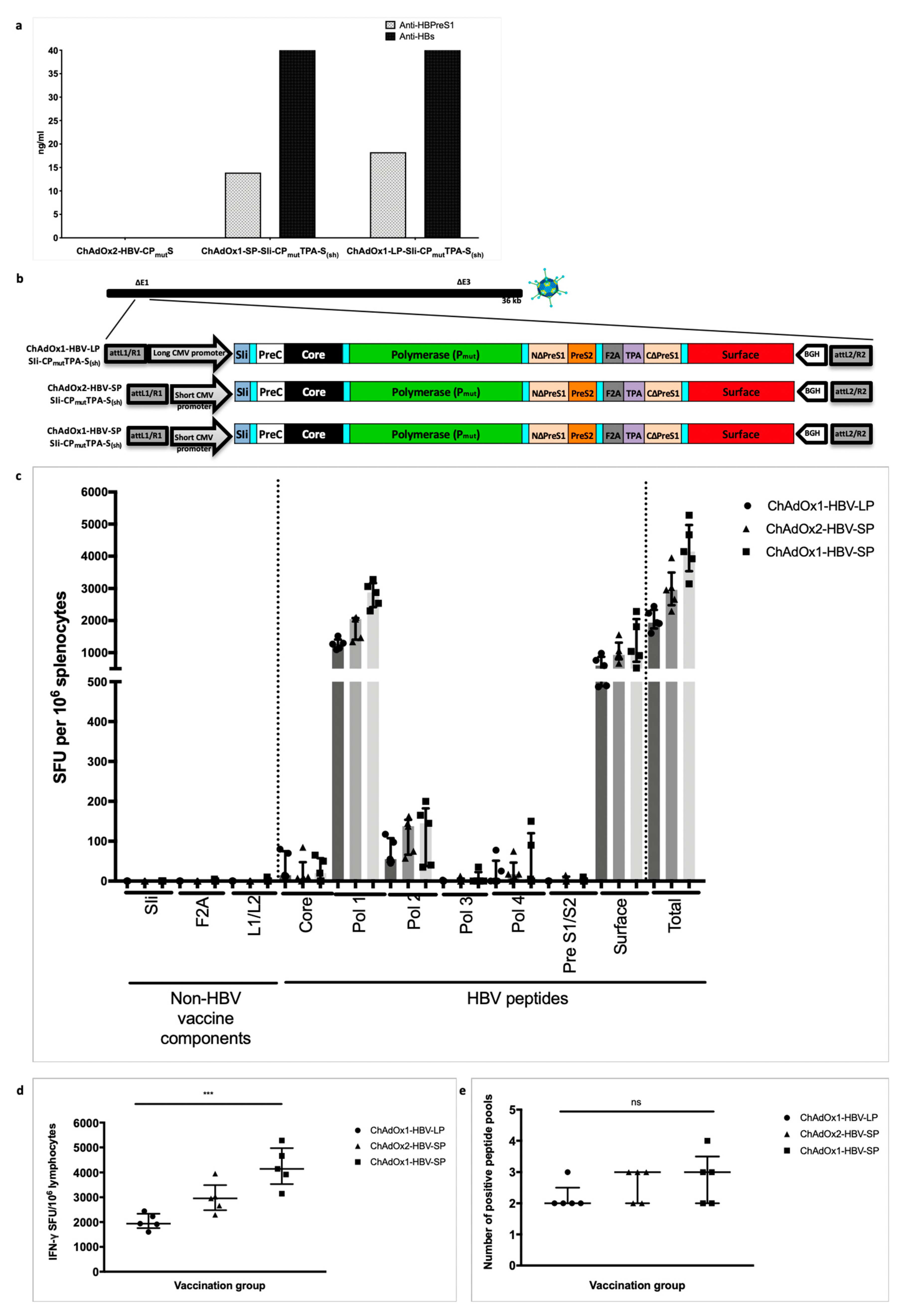
3.4. HBV Immunogen Optimisation to Promote HBsAg Secretion
3.5. Improving ChAd HBV Vaccine Stability
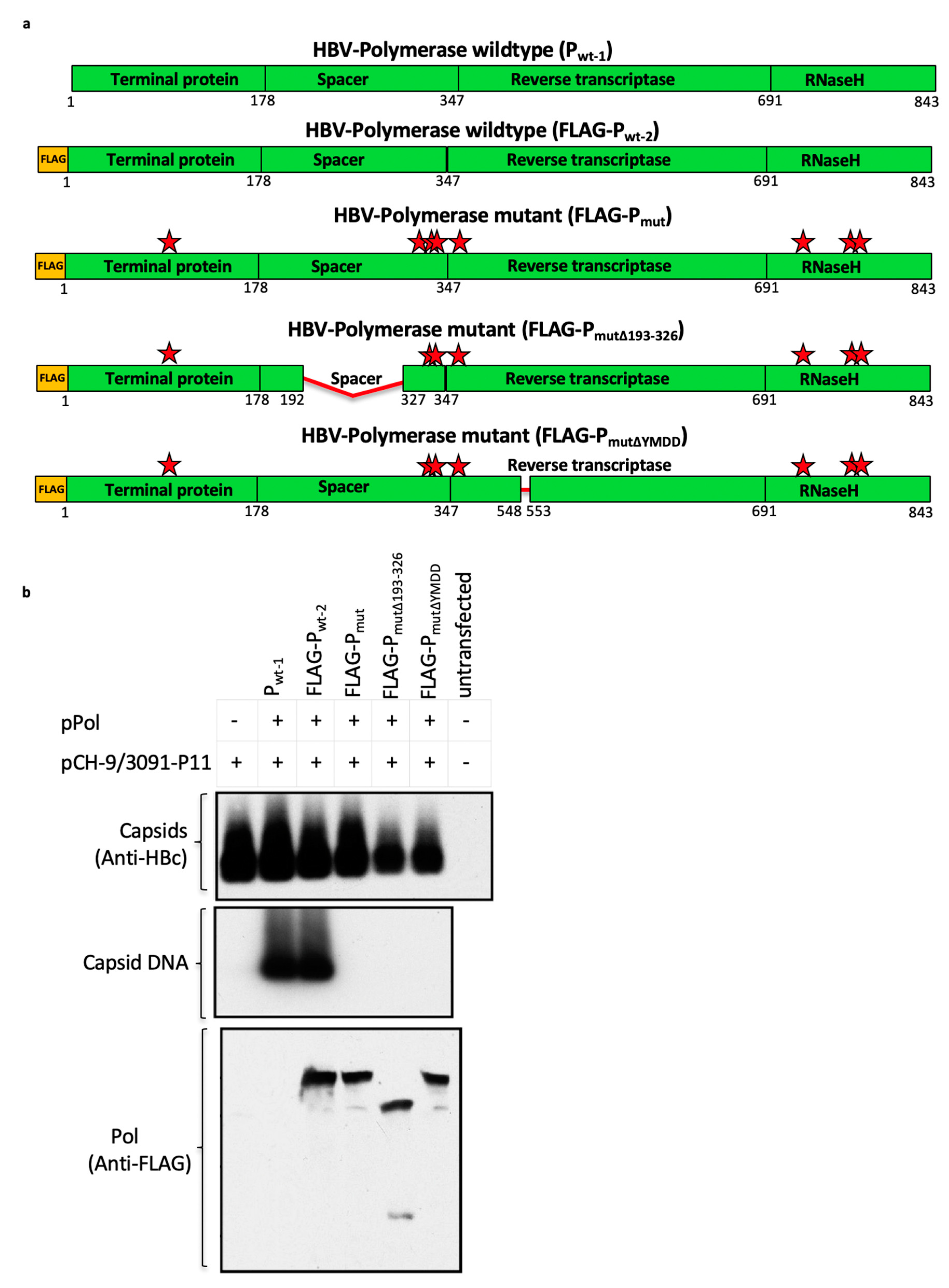
3.6. Mutant Polymerase (Pmut) Encoded within the HBV-Immunogen is Non-Functional
3.7. Vaccinating with MVA-SIi-CPmutTPA-S(sh) 7–8 weeks after ChAdOx1-SP-SIi-CPmutTPA-S(sh) Boosts the Magnitude of Vaccine-Induced T cell Responses
3.8. T cell Responses Towards HBV-Core Peptides Can Be Induced in HHD Mice
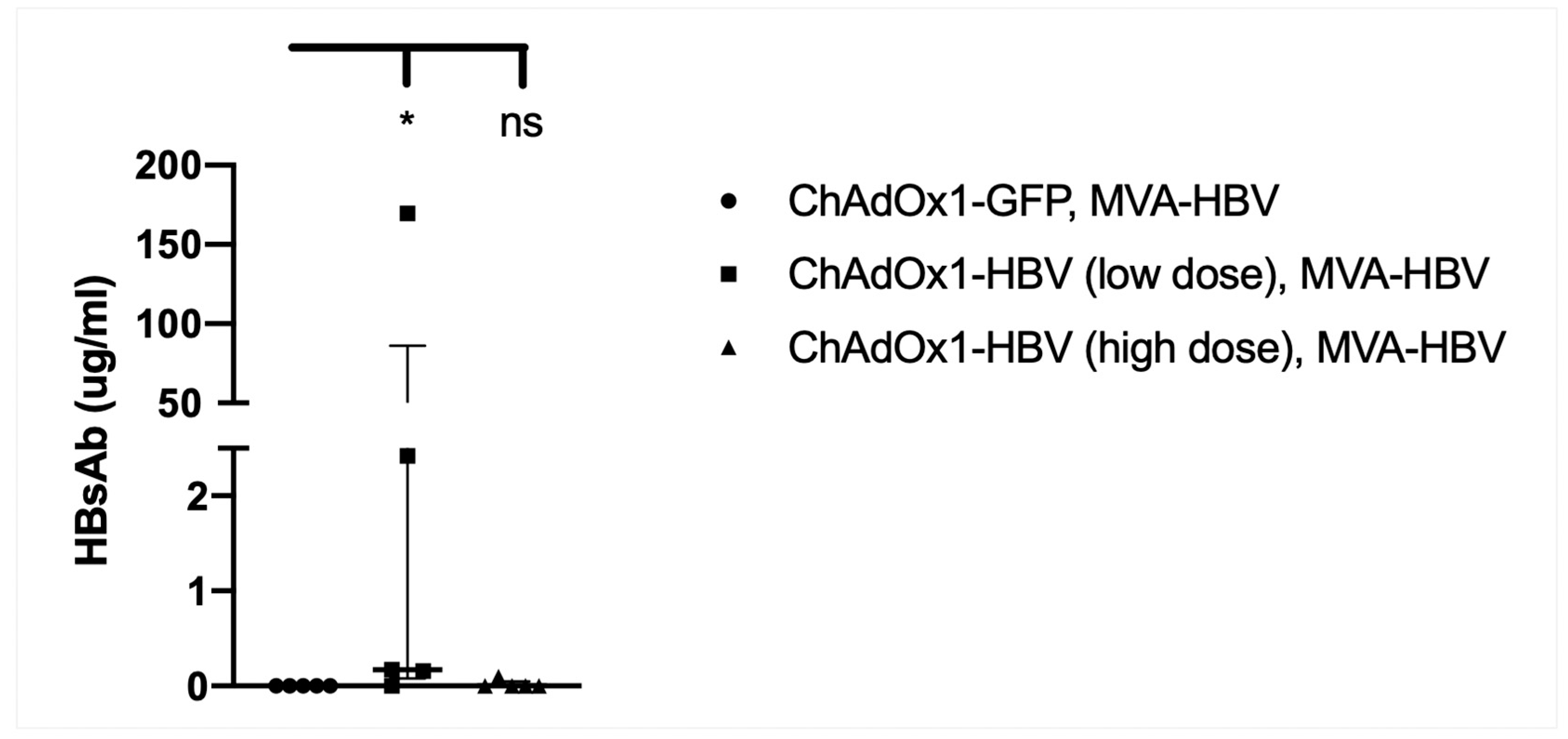
3.9. Vaccination with MVA-SIi-CPmutTPA-S(sh) 7–8 Weeks after ChAdOx1-SP-SIi-CPmutTPA-S(sh) Induces HBs-Antibody
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Nomenclature
References
- Chan, H.L.; Chan, C.K.; Hui, A.J.; Chan, S.; Poordad, F.; Chang, T.T.; Mathurin, P.; Flaherty, J.F.; Lin, L.; Corsa, A.; et al. Effects of tenofovir disoproxil fumarate in hepatitis B e antigen-positive patients with normal levels of alanine aminotransferase and high levels of hepatitis B virus DNA. Gastroenterology 2014, 146, 1240–1248. [Google Scholar] [CrossRef] [Green Version]
- Li, M.R.; Xi, H.L.; Wang, Q.H.; Hou, F.Q.; Huo, N.; Zhang, X.X.; Li, F.; Xu, X.Y. Kinetics and prediction of HBsAg loss during long-term therapy with nucleos(t)ide analogues of different potency in patients with chronic hepatitis B. PLoS ONE 2014, 9, e98476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papatheodoridis, G.V.; Chan, H.L.; Hansen, B.E.; Janssen, H.L.; Lampertico, P. Risk of hepatocellular carcinoma in chronic hepatitis B: Assessment and modification with current antiviral therapy. J. Hepatol. 2015, 62, 956–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.P.; Kao, F.Y.; Wu, C.Y.; Hung, Y.P.; Chao, Y.; Chou, Y.J.; Li, C.P. Nucleos(t)ide analogues associated with a reduced risk of hepatocellular carcinoma in hepatitis B patients: A population-based cohort study. Cancer 2015, 121, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Lin, J.T.; Ho, H.J.; Su, C.W.; Lee, T.Y.; Wang, S.Y.; Wu, C.; Wu, J.C. Association of nucleos(t)ide analogue therapy with reduced risk of hepatocellular carcinoma in patients with chronic hepatitis B: A nationwide cohort study. Gastroenterology 2014, 147, 143–151.e145. [Google Scholar] [CrossRef] [Green Version]
- Heim, K.; Neumann-Haefelin, C.; Thimme, R.; Hofmann, M. Heterogeneity of HBV-Specific CD8. Front. Immunol. 2019, 10, 2240. [Google Scholar] [CrossRef]
- Lok, A.S.; Zoulim, F.; Dusheiko, G.; Ghany, M.G. Hepatitis B cure: From discovery to regulatory approval. J. Hepatol. 2017, 67, 847–861. [Google Scholar] [CrossRef] [Green Version]
- Swadling, L.; Capone, S.; Antrobus, R.D.; Brown, A.; Richardson, R.; Newell, E.W.; Halliday, J.; Kelly, C.; Bowen, D.; Fergusson, J.; et al. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci. Transl. Med. 2014, 6, 261ra153. [Google Scholar] [CrossRef] [Green Version]
- Ewer, K.J.; O’Hara, G.A.; Duncan, C.J.; Collins, K.A.; Sheehy, S.H.; Reyes-Sandoval, A.; Goodman, A.L.; Edwards, N.J.; Elias, S.C.; Halstead, F.D.; et al. Protective CD8+ T-cell immunity to human malaria induced by chimpanzee adenovirus-MVA immunisation. Nat. Commun. 2013, 4, 2836. [Google Scholar] [CrossRef] [Green Version]
- Hayton, E.J.; Rose, A.; Ibrahimsa, U.; Del Sorbo, M.; Capone, S.; Crook, A.; Black, A.P.; Dorrell, L.; Hanke, T. Safety and tolerability of conserved region vaccines vectored by plasmid DNA, simian adenovirus and modified vaccinia virus ankara administered to human immunodeficiency virus type 1-uninfected adults in a randomized, single-blind phase I trial. PLoS ONE 2014, 9, e101591. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Mondal, M.; Zhou, D. Development of novel vaccine vectors: Chimpanzee adenoviral vectors. Hum. Vaccin. Immunother. 2018, 14, 1679–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alharbi, N.K.; Padron-Regalado, E.; Thompson, C.P.; Kupke, A.; Wells, D.; Sloan, M.A.; Grehan, K.; Temperton, N.; Lambe, T.; Warimwe, G.; et al. ChAdOx1 and MVA based vaccine candidates against MERS-CoV elicit neutralising antibodies and cellular immune responses in mice. Vaccine 2017, 35, 3780–3788. [Google Scholar] [CrossRef] [PubMed]
- Halbroth, B.R.; Sebastian, S.; Poyntz, H.C.; Bregu, M.; Cottingham, M.G.; Hill, A.V.S.; Spencer, A.J. Development of a Molecular Adjuvant to Enhance Antigen-Specific CD8(+) T Cell Responses. Sci. Rep. 2018, 8, 15020. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, S.; Bervas, N.; Ure, J.M.; Smith, A.G.; Lemonnier, F.A.; Pérarnau, B. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. J. Exp. Med. 1997, 185, 2043–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- du Sert, N.P.; Bamsey, I.; Bate, S.T.; Berdoy, M.; Clark, R.A.; Cuthill, I.C.; Fry, D.; Karp, N.A.; Macleod, M.; Moon, L.; et al. The Experimental Design Assistant. Nat. Methods 2017, 14, 1024–1025. [Google Scholar] [CrossRef] [Green Version]
- Percie du Sert, N.; Bamsey, I.; Bate, S.T.; Berdoy, M.; Clark, R.A.; Cuthill, I.; Fry, D.; Karp, N.A.; Macleod, M.; Moon, L.; et al. The Experimental Design Assistant. PLoS Biol. 2017, 15, e2003779. [Google Scholar] [CrossRef] [Green Version]
- Rehermann, B.; Fowler, P.; Sidney, J.; Person, J.; Redeker, A.; Brown, M.; Moss, B.; Sette, A.; Chisari, F.V. The cytotoxic T lymphocyte response to multiple hepatitis B virus polymerase epitopes during and after acute viral hepatitis. J. Exp. Med. 1995, 181, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Shimada, N.; Yamamoto, K.; Kuroda, M.J.; Terada, R.; Hakoda, T.; Shimomura, H.; Hata, H.; Nakayama, E.; Shiratori, Y. HBcAg-specific CD8 T cells play an important role in virus suppression, and acute flare-up is associated with the expansion of activated memory T cells. J. Clin. Immunol. 2003, 23, 223–232. [Google Scholar] [CrossRef]
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 2003, 77, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Koh, S.S.; Lee, C.G. Hepatitis B Virus X Protein and Hepatocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 940. [Google Scholar] [CrossRef] [Green Version]
- HBVdb, a HBV database. Available online: https://hbvdb.ibcp.fr/HBVdb/ (accessed on 26 August 2015).
- Hayer, J.; Jadeau, F.; Deleage, G.; Kay, A.; Zoulim, F.; Combet, C. HBVdb: A knowledge database for Hepatitis B Virus. Nucleic Acids Res. 2013, 41, D566–D570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MAFFT, a multiple sequence alignment program. Available online: https://mafft.cbrc.jp/alignment/server/ (accessed on 26 August 2015).
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Cao, G. Genotypes, mutations, and viral load of hepatitis B virus and the risk of hepatocellular carcinoma: HBV properties and hepatocarcinogenesis. Hepat. Mon. 2011, 11, 86–91. [Google Scholar] [PubMed]
- Morris, S.J.; Sebastian, S.; Spencer, A.J.; Gilbert, S.C. Simian adenoviruses as vaccine vectors. Future Virol. 2016, 11, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Wen, B.; Deng, Y.; Guan, J.; Yan, W.; Wang, Y.; Tan, W.; Gao, J. Signal peptide replacements enhance expression and secretion of hepatitis C virus envelope glycoproteins. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Dicks, M.D.; Spencer, A.J.; Edwards, N.J.; Wadell, G.; Bojang, K.; Gilbert, S.C.; Hill, A.V.; Cottingham, M.G. A novel chimpanzee adenovirus vector with low human seroprevalence: Improved systems for vector derivation and comparative immunogenicity. PLoS ONE 2012, 7, e40385. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Clark, D.N.; Cao, F.; Tavis, J.E.; Hu, J. Comparative analysis of hepatitis B virus polymerase sequences required for viral RNA binding, RNA packaging, and protein priming. J. Virol. 2014, 88, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Hu, J. Hepatitis B virus reverse transcriptase: Diverse functions as classical and emerging targets for antiviral intervention. Emerg. Microbes Infect. 2013, 2, e56. [Google Scholar] [CrossRef]
- Ko, C.; Shin, Y.C.; Park, W.J.; Kim, S.; Kim, J.; Ryu, W.S. Residues Arg703, Asp777, and Arg781 of the RNase H domain of hepatitis B virus polymerase are critical for viral DNA synthesis. J. Virol. 2014, 88, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M. Hepatitis B viruses: Reverse transcription a different way. Virus Res. 2008, 134, 235–249. [Google Scholar] [CrossRef]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Sandoval, A.; Berthoud, T.; Alder, N.; Siani, L.; Gilbert, S.C.; Nicosia, A.; Colloca, S.; Cortese, R.; Hill, A.V. Prime-boost immunization with adenoviral and modified vaccinia virus Ankara vectors enhances the durability and polyfunctionality of protective malaria CD8+ T-cell responses. Infect. Immun. 2010, 78, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Sandoval, A.; Wyllie, D.H.; Bauza, K.; Milicic, A.; Forbes, E.K.; Rollier, C.S.; Hill, A.V. CD8+ T effector memory cells protect against liver-stage malaria. J. Immunol. 2011, 187, 1347–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tully, C.M.; Chinnakannan, S.; Mullarkey, C.E.; Ulaszewska, M.; Ferrara, F.; Temperton, N.; Gilbert, S.C.; Lambe, T. Novel Bivalent Viral-Vectored Vaccines Induce Potent Humoral and Cellular Immune Responses Conferring Protection against Stringent Influenza A Virus Challenge. J. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, L.; Sridhar, S.; Payne, R.; Edmans, M.; Milicic, A.; Venkatraman, N.; Lugonja, B.; Clifton, L.; Qi, C.; Folegatti, P.M.; et al. Heterologous Two-Dose Vaccination with Simian Adenovirus and Poxvirus Vectors Elicits Long-Lasting Cellular Immunity to Influenza Virus A in Healthy Adults. EBioMedicine 2018, 29, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Ewer, K.; Rampling, T.; Venkatraman, N.; Bowyer, G.; Wright, D.; Lambe, T.; Imoukhuede, E.B.; Payne, R.; Fehling, S.K.; Strecker, T.; et al. A Monovalent Chimpanzee Adenovirus Ebola Vaccine Boosted with MVA. N. Engl. J. Med. 2016, 374, 1635–1646. [Google Scholar] [CrossRef]
- Green, C.A.; Sande, C.J.; Scarselli, E.; Capone, S.; Vitelli, A.; Nicosia, A.; Silva-Reyes, L.; Thompson, A.J.; de Lara, C.M.; Taylor, K.S.; et al. Novel genetically-modified chimpanzee adenovirus and MVA-vectored respiratory syncytial virus vaccine safely boosts humoral and cellular immunity in healthy older adults. J. Infect. 2019, 78, 382–392. [Google Scholar] [CrossRef]
- Green, C.A.; Scarselli, E.; Sande, C.J.; Thompson, A.J.; de Lara, C.M.; Taylor, K.S.; Haworth, K.; Del Sorbo, M.; Angus, B.; Siani, L.; et al. Chimpanzee adenovirus- and MVA-vectored respiratory syncytial virus vaccine is safe and immunogenic in adults. Sci. Transl. Med. 2015, 7, 300ra126. [Google Scholar] [CrossRef] [Green Version]
- Mensah, V.A.; Gueye, A.; Ndiaye, M.; Edwards, N.J.; Wright, D.; Anagnostou, N.A.; Syll, M.; Ndaw, A.; Abiola, A.; Bliss, C.; et al. Safety, Immunogenicity and Efficacy of Prime-Boost Vaccination with ChAd63 and MVA Encoding ME-TRAP against Plasmodium falciparum Infection in Adults in Senegal. PLoS ONE 2016, 11, e0167951. [Google Scholar] [CrossRef] [Green Version]
- Ghozy, S.; Nam, N.H.; Radwan, I.; Karimzadeh, S.; Tieu, T.M.; Hashan, M.R.; Abbas, A.S.; Eid, P.S.; Vuong, N.L.; Khang, N.V.; et al. Therapeutic efficacy of hepatitis B virus vaccine in treatment of chronic HBV infections: A systematic review and meta-analysis. Rev. Med. Virol. 2019, e2089. [Google Scholar] [CrossRef]
- Bliss, C.M.; Bowyer, G.; Anagnostou, N.A.; Havelock, T.; Snudden, C.M.; Davies, H.; de Cassan, S.C.; Grobbelaar, A.; Lawrie, A.M.; Venkatraman, N.; et al. Assessment of novel vaccination regimens using viral vectored liver stage malaria vaccines encoding ME-TRAP. Sci. Rep. 2018, 8, 3390. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Boni, C.; Ogg, G.S.; King, A.S.; Reignat, S.; Lee, C.K.; Larrubia, J.R.; Webster, G.J.; McMichael, A.J.; Ferrari, C.; et al. Direct ex vivo analysis of hepatitis B virus-specific CD8(+) T cells associated with the control of infection. Gastroenterology 1999, 117, 1386–1396. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, E.; Ma, Z.; Wu, W.; Kosinska, A.; Zhang, X.; Moller, I.; Seiz, P.; Glebe, D.; Wang, B.; et al. Enhancing virus-specific immunity in vivo by combining therapeutic vaccination and PD-L1 blockade in chronic hepadnaviral infection. PLoS Pathog. 2014, 10, e1003856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atcheson, E.; Li, W.; Bliss, C.M.; Chinnakannan, S.; Heim, K.; Sharpe, H.; Hutchings, C.; Dietrich, I.; Nguyen, D.; Kapoor, A.; et al. Use of an Outbred Rat Hepacivirus Challenge Model for Design and Evaluation of Efficacy of Different Immunization Strategies for Hepatitis C Virus. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chinnakannan, S.K.; Cargill, T.N.; Donnison, T.A.; Ansari, M.A.; Sebastian, S.; Lee, L.N.; Hutchings, C.; Klenerman, P.; Maini, M.K.; Evans, T.; et al. The Design and Development of a Multi-HBV Antigen Encoded in Chimpanzee Adenoviral and Modified Vaccinia Ankara Viral Vectors; A Novel Therapeutic Vaccine Strategy against HBV. Vaccines 2020, 8, 184. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines8020184
Chinnakannan SK, Cargill TN, Donnison TA, Ansari MA, Sebastian S, Lee LN, Hutchings C, Klenerman P, Maini MK, Evans T, et al. The Design and Development of a Multi-HBV Antigen Encoded in Chimpanzee Adenoviral and Modified Vaccinia Ankara Viral Vectors; A Novel Therapeutic Vaccine Strategy against HBV. Vaccines. 2020; 8(2):184. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines8020184
Chicago/Turabian StyleChinnakannan, Senthil K., Tamsin N. Cargill, Timothy A. Donnison, M. Azim Ansari, Sarah Sebastian, Lian Ni Lee, Claire Hutchings, Paul Klenerman, Mala K. Maini, Tom Evans, and et al. 2020. "The Design and Development of a Multi-HBV Antigen Encoded in Chimpanzee Adenoviral and Modified Vaccinia Ankara Viral Vectors; A Novel Therapeutic Vaccine Strategy against HBV" Vaccines 8, no. 2: 184. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines8020184