

Abstract
Objective. To investigate the effects of soluble uric acid (UA) on expression and activation of the NOD-like receptor (NLR) pyrin domain containing protein 3 (NLRP3) inflammasome in human monocytes to elucidate the role of hyperuricemia in the pathogenesis of gout.
Methods. Primary human monocytes and the THP-1 human monocyte cell line were used to determine the effects of short- and longterm exposure to UA on activation of the NLRP3 inflammasome and subsequent interleukin 1β (IL-1β) secretion by ELISA and cell-based assays. Expression of key NLRP3 components in monocytes from patients with a history of gout were analyzed by quantitative PCR.
Results. Precipitation of UA was required for activation of the NLRP3 inflammasome and subsequent release of IL-1β in human monocytes. Neither monosodium urate (MSU) crystals nor soluble UA had any effect on activation of the transcription factor, nuclear factor-κB. Prolonged exposure of monocytes to soluble UA did not alter these responses. However, both MSU crystals and soluble UA did result in a 2-fold increase in reactive oxygen species. Patients with gout (n = 15) had significantly elevated serum UA concentrations compared to healthy individuals (n = 16), yet secretion of IL-1β and expression of NLRP3 inflammasome components in monocytes isolated from these patients were not different from those of healthy controls.
Conclusion. Despite reports indicating that soluble UA can prime and activate the NLRP3 inflammasome in human peripheral blood mononuclear cells, precipitation of soluble UA into MSU crystals is essential for in vitro NLRP3 signaling in primary human monocytes.
Uric acid (UA) is a major antioxidant in the plasma that can help protect against oxidative stress induced by free radicals. Indeed, this function of UA is thought to have led to the evolutionary loss of the uricase gene in humans, resulting in much higher circulating concentrations of UA in humans than in other mammals1,2. Clinically relevant hyperuricemia is defined as a plasma UA concentration in excess of 6.8 mg/dl3, resulting in increased risk of precipitation to form potentially proinflammatory monosodium urate (MSU) crystals4.
Hyperuricemia is associated with several pathological conditions including metabolic syndrome5, hypertension6, chronic kidney disease7, and cardiovascular disease8. It is also the biggest single risk factor for gout, the pathophysiology of which is primarily driven by the production of the proinflammatory cytokine interleukin 1β (IL-1β), released as a result of recognition of MSU crystals by mononuclear cells9. Intriguingly, only about 10% of hyperuricemic individuals ever develop gout10, nor does the presence of MSU crystals within joints necessarily precipitate a gout attack11.
In vivo processing and subsequent secretion of IL-1β occurs through activation of the NOD-like receptor (NLR) pyrin domain containing protein 3 (NLRP3) inflammasome. This is a tripartite cytosolic complex formed of 3 proteins: NLRP3, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and pro-caspase-1. Upon activation, NLRP3 oligomerizes with the adaptor protein ASC, which in turn mediates the recruitment of the inactive zymogen pro-caspase-112. Oligomerization of inflammasome components ultimately results in the autoproteolytic cleavage of pro-caspase-1 into its active form. Caspase-1 then cleaves pro-IL-1β and pro-IL-18 into their active forms, resulting in their secretion from cells13.
It is generally accepted that activation of the NLRP3 inflammasome requires 2 distinct signals, although an alternative pathway has been identified in human monocytes in response to lipopolysaccharide (LPS)14. These 2 signals consist of a priming signal to induce transcription of both NLRP3 and pro-IL-1β, and a second signal that induces oligomerization of the inflammasome. Various ligands can induce NLRP3 priming, including the toll-like receptor (TLR) 2 ligand Pam3CSK4 (Pam3) and the TLR4 ligand LPS through activation of nuclear factor-κB (NF-κB)15. Several structurally diverse stimuli can function as the second signal, but none have been found to interact directly with inflammasome proteins16,17. Possible mechanisms by which second signals activate the inflammasome include lysosomal degradation18, cationic flux19, and generation of reactive oxygen species (ROS)20.
Despite hyperuricemia being the single greatest risk factor for development of gout, the precise role of UA in its pathophysiology remains to be elucidated. Studies have shown that soluble UA can influence both the priming and activation of the inflammasome even in the absence of MSU crystal formation21,22,23. However, many of these studies were completed in human peripheral blood mononuclear cells (PBMC) or mouse bone marrow-derived macrophages, which may not accurately reflect the responses of primary human monocytes.
It is important to fully define the role of UA in activating the NLRP3 inflammasome in these cells, because monocytes rather than PBMC are the major producers of IL-1β during an acute attack of gout24. Thus our aim was to determine the effect of soluble UA on activation of NLRP3 in human monocytes to investigate how hyperuricemia can increase susceptibility to gout at the cellular level. We used a combination of human monocytic THP-1 cells and primary human monocytes from healthy controls and patients with gout to investigate the acute and longer-term effects of UA on expression, priming, and activation of the NLRP3 inflammasome.
MATERIALS AND METHODS
Reagents
Cell culture media, fetal bovine serum, and uric acid were purchased from Sigma-Aldrich. Opti-MEM media and penicillin/streptomycin were purchased from Thermo Fisher Scientific. MSU crystals, MCC95025, and QUANTI-Blue reagent were purchased from Invivogen. Qiazol, QuantiTect Reverse Transcription kit, and the QuantiFast-SYBR green PCR kit were from Qiagen.
Isolation and stimulation of primary human monocytes
Single-donor platelet phoresis residues were obtained from the North London Blood Transfusion Centre. PBMC were isolated by density gradient separation using lympholyte-H cell separation media (VH Bio) followed by isolation of monocytes by Percoll (Sigma-Aldrich) density gradient centrifugation26. Cells were stored in liquid nitrogen for future use.
The appropriate concentration of MSU crystals for stimulating cells was determined by titration to achieve activation of NLRP3 with no effects on viability (data not shown). Soluble UA concentrations were chosen to allow direct comparison with MSU crystal concentrations and were subsequently increased to represent a wider range of hyperuricemia. UA was dissolved in prewarmed RPMI at a final concentration of 0.6–1.0 mg/ml and filter-sterilized using 0.20 µm filters. Crystals were not detectable in preparations of uric acid, nor were crystals ever observed under any of the experimental conditions used (Supplementary Figure 1, available with the online version of this article). For experiments, monocytes were resuspended in RPMI cell culture media containing 5% fetal calf serum (FCS) and seeded at 3 × 105 cells/well in 96-well plates. For experiments involving preincubation of cells with soluble UA, cells were allowed to adhere for 2 h and then incubated in RPMI (5% FCS) ± 30 mg/dl UA for 10–16 h. Cells were then washed with RPMI and stimulants added for the specified time.
For some experiments, monocytes were isolated from 30 ml blood collected from patients with gout (n = 15) or healthy volunteer controls (n = 16). Patients were defined as those who had had an inflammatory arthritic attack clinically diagnosed as gout. The majority (10/16) were prescribed antiinflammatory medication, with 50% prescribed allopurinol. PBMC were isolated by density gradient separation using lympholyte-H followed by purification of monocytes using CD14+ magnetic bead– (Miltenyi Biotec) positive isolation and stored in liquid nitrogen until use. Matched serum samples were collected from each donor and stored at −80°C. For experiments, cells were seeded at 4 × 104 cells/well in 384-tissue culture plates and allowed to adhere for 2 h, then stimulated for 18 h in culture media containing Pam3 ± MSU.
Culture and stimulation of THP-1 cells
The human THP-1 monocytic cell line was cultured in RPMI-1640 medium supplemented with 10% (vol/vol) FCS and streptomycin/penicillin (100 µg/ml/100 U/ml, respectively). THP-1 cells were seeded at 1.6 × 105 cells/well in 96-well plates and stimulated 18 h with Pam3, UA, or MSU before cell-conditioned media were collected for analyses. The THP1-Blue cell line (Invivogen, Toulouse, France) is derived from human THP-1 monocytes with stable integration of a NF-κB–inducible secreted embryonic alkaline phosphatase (SEAP) reporter construct and cultured as described for THP-1 cells with the addition of 100 µg/ml of Zeocin to maintain the reporter plasmid. For experiments requiring preincubation with UA, cells were cultured in the presence of 30 mg/dl of UA in complete RPMI for 24–48 h and then seeded and stimulated for the specified times.
Measurement of NF-κB activation in THP1-Blue cells
THP1-Blue cells were stimulated for 20 h with 100 ng/ml Pam3, 10–40 mg/dl UA, and/or 10–20 mg/dl MSU crystals. SEAP levels were determined using Quanti-Blue assay (Invivogen), according to the manufacturer’s instructions.
Quantification of IL-1β
IL-1β in cell supernatants was measured by ELISA using matched anti-human IL-1β antibodies (R&D Systems).
ROS measurements
ROS generation was detected using ROS-Glo assay (Promega), according to the manufacturer’s instructions27. Briefly, cells were stimulated 2 h with 100 ng/ml Pam3, 10 mg/dl MSU, or 30 mg/dl UA in the presence of 25 µM ROS-Glo H2O2 substrate and ROS levels were determined by the addition of ROS-Glo detection reagent. Luminescence was measured by spectrophotometry.
Measurement of serum UA
Serum UA concentrations were measured using an Amplex Red–based uric acid/uricase assay kit (ThermoFisher), according to the manufacturer’s instructions. Fluorescence of Amplex Red reagent was measured at ex530/em590 nm.
RNA extraction, reverse transcription, and absolute quantitative real-time PCR (qRT-PCR)
Monocytes were washed twice in phosphate buffered saline, then RNA was extracted (RNeasy kit, Qiagen) and reverse transcribed to cDNA (QuantiTect reverse transcription kit, Qiagen) according to the manufacturer’s instructions. qRT-PCR assays for absolute quantification of gene expression were purchased from qStandard. Copy numbers for NLRP3, CASP1, IL1β, and PYCARD (gene name for ASC) were determined by qRT-PCR (QuantiFast SYBR Green PCR kit, Qiagen) on a Stratagene Mx3000 thermocycler (Agilent Technologies) or Rotor-Gene thermocycler (Qiagen) under the following thermocycling program: 40 cycles of 95°C for 15 s and 60°C for 30 s with an initial cycle of 95°C for 15 min. The following primers were used: human β2-microglobulin (B2M), forward 5′-CTC TCT CTT TCT GGC CTG GAG-3′ and reverse 5′-ACC CAG ACA CAT AGC AAT TCA G-3′; human ribosomal protein L32 (RPL32), forward 5′-CAT CTC CTT CTC GGC ATC AT-3′ and reverse 5′-ACC CTG TTG TCA ATG CCT CT-3′; human CASP1, forward 5′-ATG CCT GTT CCT GTG ATG TGG-3′ and reverse 5′-CTC TTC ACT TCC TGC CCA CAG A-3′; human IL1B, forward 5′-GTA ATG ACA AAA TAC CTG TGG CCT TG-3′ and reverse 5′-TTT GGG ATC TAC ACT CTC CAG C-3′; human NLRP3, forward 5′-GAG ATG AGC CGA AGT GGG GTT C-3′ and reverse 5′-GCT TCT CAC GTA CTT TCT GTA CTT CT-3′; human PYCARD, forward 5′-GCT AAC GTG CTG CGC GAC AT-3′ and reverse 5′-CCA CTC AAC GTT TGT GAC CCT-3′. Copy number for each gene of interest and reference gene was calculated by interpolation from a standard curve, range 101–107 copies, run on the same plate. B2M and RPL32 were used as reference genes.
Statistical analysis
Differences between groups were assessed using unpaired t tests, Mann-Whitney U tests, or 1-way ANOVA followed by Tukey’s multiple comparison test after testing for differences in variation using the Brown-Forsythe test. Correlation analysis was performed using Pearson’s correlation coefficient. The level of significance was set at p < 0.05. All analyses were conducted using GraphPad Prism v. 7 (GraphPad).
Ethics statement
The UK National Research Ethics Service approval (NRES ref. 15/NS/0083) was obtained for collection of human blood samples with informed written consent. No patient-identifiable information was accessed by researchers. For purchased blood products, local ethical approval was given by the Brighton and Sussex Medical School Research Governance and Ethics Committee (R&D ref. 15/130/MUL).
RESULTS
Effect of MSU and soluble UA on activation of NLRP3 inflammasome in human monocytes
THP-1 monocytes or primary human monocytes were used to investigate activation of the NLRP3 inflammasome in response to MSU crystals or soluble UA. The TLR2 ligand, Pam3, was used as a priming signal and secretion of IL-1β used as a measure of NLRP3 activation. To confirm the activation of NLRP3, cells were stimulated in the presence of MCC950, a chemical inhibitor of NLRP3. In both THP-1 monocytes (Supplementary Figure 2A, available with the online version of this article) and primary human monocytes (Supplementary Figure 2B), MCC950 inhibited IL-1β secretion in response to MSU crystals by 90%, confirming NLRP3-dependent secretion. Small increases in secretion of IL-1β were observed in response to Pam3 alone, with significantly greater release of IL-1β in the presence of MSU in both THP-1 monocytes (Figure 1A) and primary human monocytes (Figure 1C). In contrast, addition of soluble UA did not increase Pam3-induced IL-1β secretion in either THP-1 cells (Figure 1B) or primary human monocytes (Figure 1D). THP-1-Blue cells were used to investigate whether MSU or soluble UA had any effect on priming of the NLRP3 pathway. Neither MSU (Figure 1E) nor soluble UA (Figure 1F) treatment alone had any effect on activation of NF-κB in these cells, although the higher concentration of MSU did increase Pam3-induced NF-κB.

Priming and activation of the NLRP3 inflammasome in human monocytes. THP-1 (A, B) or primary human monocytes (C, D) were stimulated for 18 h with Pam3 ± MSU crystals or soluble UA. IL-1β secretion was determined by ELISA. THP1-Blue monocytes (E, F) were stimulated for 20 h with Pam3, soluble UA, or MSU crystals. Nuclear factor-κB (NF-κB) activation was determined with QUANTI-Blue assay. Data represent means from 3–4 separate experiments (THP-1 and THP1-Blue cells) or 3–5 individual donors (primary monocytes) ± SEM. Significance by 1-way ANOVA or unpaired t test (A, B). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. NLRP3: NOD-like receptor pyrin domain containing protein 3; MSU: monosodium urate; UA: uric acid; IL-1β: interleukin 1β; SEM: standard error of the mean; SEAP: secreted embryonic alkaline phosphatase.
In hyperuricemic individuals, circulating monocytes are exposed to elevated concentrations of soluble UA for prolonged periods. We therefore hypothesized that monocytes exposed to soluble UA prior to stimulation could have altered priming and activation of the NLRP3 inflammasome. To test this, primary human and THP-1 monocytes were exposed to soluble UA (30 mg/dl) for up to 48 h. This concentration of soluble UA is well in excess of the accepted range for hyperuricemia (> 6.8 mg/dl). Longer incubation times were tested in THP-1 cells compared with primary human monocytes because of decreases in viability in primary cells after 24 h culture. Preincubation of primary human monocytes with soluble UA for 10 h (Figure 2A), or THP-1 cells for 24–48 h (Figure 2B, 2C) had no effect on the release of IL-1β from these cells. Further, activation of NF-κB in response to Pam3 ± MSU was also unaffected by preincubation with soluble UA for 24 h (Supplementary Figure 3A, 3B, available with the online version of this article) or 48 h (Figure 3D, 3E).

Effect of preincubation with soluble UA on priming and activation of NLRP3 in human monocytes. (A) Primary human monocytes were incubated with RPMI or soluble UA (30 mg/dl) for 10 h prior to treatments indicated for 12 h. THP-1 monocytes were incubated with soluble UA for 24 h (B) or 48 h (C) prior to indicated treatments for 18 h. IL-1β secretion was determined by ELISA. THP1-Blue monocytes were pre-exposed to RPMI or soluble UA for 48 h (D, E), followed by 20 h stimulation. NF-κB activation was determined by QUANTI-Blue assay. Data represent means ± SEM for 4 separate donors or 3–4 separate experiments. Significance determined by unpaired t test. NLRP3: NOD-like receptor pyrin domain containing protein 3; MSU: monosodium urate; UA: uric acid; IL-1β: interleukin 1β; SEM: standard error of the mean; SEAP: secreted embryonic alkaline phosphatase; NF-κB: nuclear factor-κB.

Production of ROS in primary human monocytes. Primary human monocytes were treated with catalase ± MSU crystals for 2 h (A) or MSU 10 mg/dl (B) for 6 h and ROS levels were measured by ROS-Glo assay. Primary human monocytes were exposed to Pam3 ± MSU or soluble UA for 2 h (C) or for 2 h following 16 h preincubation (D) in RPMI or soluble UA. ROS generation was measured by ROS-Glo luminescent assay. Data represent mean of triplicate measures from a single donor (A, B) or mean ± SEM from 4–6 separate donors (C, D). Significance determined by 1-way ANOVA or unpaired t-test. MSU: monosodium urate; ROS: reactive oxygen species; UA: uric acid; SEM: standard error of the mean; RLU: relative light units.
Induction of ROS by soluble UA and MSU crystals in primary human monocytes
To assess whether MSU crystals or soluble UA can induce generation of ROS, we measured ROS levels in human monocytes. Initial experiments used catalase to decrease ROS, or MSU to increase ROS, to confirm that the ROS-Glo assay could indeed measure changes in ROS (Figure 3A) and to determine the optimal timing for measurement of ROS in these cells (Figure 3B). Short-term (2 h) treatment with both MSU and soluble UA increased ROS generation in primary human monocytes (Figure 3C), an effect that was not seen when cells were preincubated with 30 mg/dl of soluble UA for 16 h prior to stimulation with Pam3 ± MSU or UA (Figure 3D).
Activation of NLRP3 inflammasome in monocytes isolated from patients with gout
Susceptibility to gout attacks could be related to increased cellular sensitivity to MSU crystals. To test this hypothesis, monocytes were isolated from the blood of individuals with a history of gout attacks and stimulated for 18 h with Pam3 ± MSU crystals. Serum UA concentration was significantly higher in patients with gout (n = 15) compared to healthy controls (n = 16; Figure 4A), although only 27% of the subjects with gout were clinically hyperuricemic at the time of sampling.

Serum UA concentration and stimulation of monocytes from patients with gout or healthy controls. (A) Uric acid concentration in serum was measured by Amplex Red uric acid assay. Monocytes were isolated from whole blood and exposed to Pam3 (B) or Pam3 + MSU crystals (C). IL-1β secretion was determined by ELISA. IL-1β secretion in response to MSU was analyzed by normalization to secretion induced by Pam3 alone (D). Data represent means from 3–6 repeats for each separate donor. Error bars represent average of all donors ± SEM. Significance analyzed by 2-tailed unpaired Mann-Whitney U test; *p < 0.05. UA: uric acid; MSU: monosodium urate; Pam3CSK4: Pam3; IL-1β: interleukin 1β; SEM: standard error of the mean.
There were no significant differences in IL-1β secretion between monocytes from patients with gout and controls (Figure 4B, 4C). However, there was a trend toward increased responsiveness to MSU in monocytes from patients with gout when the IL-1β secretion data were normalized to secretion induced by Pam3 alone for each donor (Figure 4D). However, IL-1β secretion from these cells did not correlate with serum UA concentration (Supplementary Figure 4, available with the online version of this article). To determine whether this trend could be explained by increased expression of genes coding for components of the NLRP3 inflammasome, qRT-PCR was used to determine constitutive expression of caspase-1 (CASP1), pro-IL-1β (IL1B), ASC (PYCARD), and NLRP3 in the absence of a stimulus. There were no differences in the expression of any of the genes tested between controls and patients (Figure 5A–5D). Caspase-1 and pro-IL-1β transcripts were expressed at 10-fold greater levels than the NLRP3 transcripts. Finally, there was no correlation between serum UA concentration and expression of the inflammasome genes measured (Figure 5D–5F) in either controls or patients.
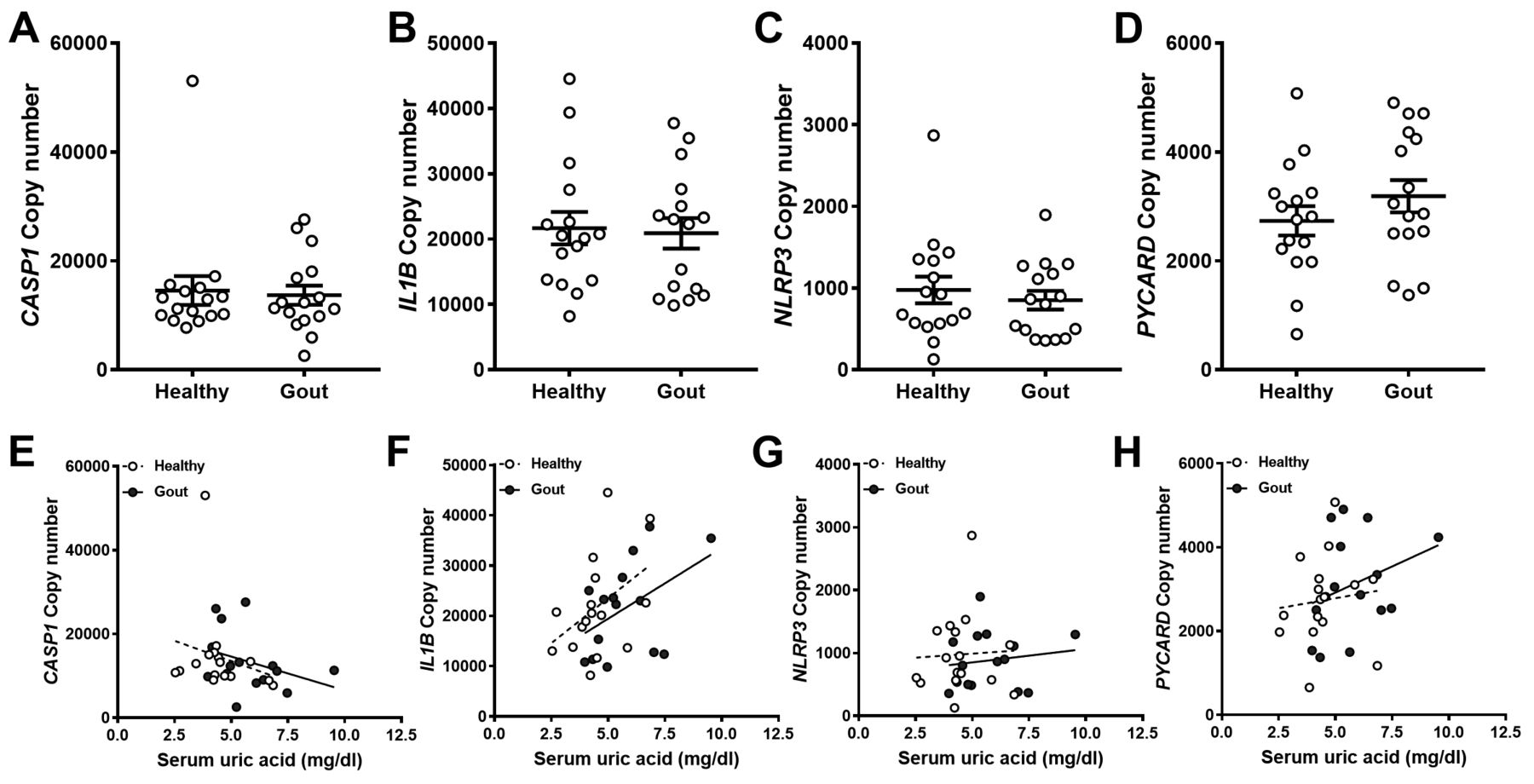
Expression of NLRP3 inflammasome components in monocytes from gout patients or healthy controls. cDNA was prepared from human monocytes, which were isolated from whole blood. Expression of CASP1 (A), IL1B (B), NLRP3 (C), and PYCARD (D) analyzed by qRT-PCR. Absolute copy numbers were calculated using standard curves for each gene of interest and normalized to expression of β2-microglobulin (β2m) and RPL32. Each point represents an average of 2–3 replicates. Error bars are derived from mean of all samples ± SEM. Significance analyzed by 2-tailed unpaired Mann-Whitney U test or Spearman correlation coefficient analysis. NLRP3: NOD-like receptor pyrin domain containing protein 3; qRT-PCR: quantitative real-time PCR; RPL32: human ribosomal protein L32; SEM: standard error of the mean.
DISCUSSION
There is a limited number of studies assessing the effects of UA in primary human cells, but the available data suggest that any effect of UA is cell-type–specific and much more subtle than the well-documented effects of crystallized UA. Thus, UA had no effect on release of IL-1β or tumor necrosis factor from human monocyte-derived macrophages, but at concentrations of 50 mg/dl did induce secretion of IL-1β from PBMC22. However, these results were not recapitulated in monocytes, where incubation with UA had no effect on IL-1β, as in the results we observed, but did increase IL-1β production when cells were stimulated with LPS ± MSU after the initial incubation with UA23. Clearly, the responses of isolated monocytes are different from those of PBMC, which is a mixed population of monocytes and lymphocytes that contains on average about 8%–10% monocytes. The differences in responses to UA between human monocytes and PBMC cannot be explained simply by differences in expression of NLRP3, because this is substantially lower in lymphocytes than in monocytes28. However, as previously shown, UA can activate T cells in an antigen-independent manner29. Perhaps there are interactions between activated T cells and monocytes that cause the monocytes in the PBMC mixture to respond to UA by secreting IL-1β, an effect not seen in pure monocyte cultures. This possibility deserves investigation and may provide insight into the activation of monocytes during hyperuricemia.
The monocytes from patients with gout studied here showed a tendency to produce more IL-1β as a result of treatment with MSU. Serum UA concentration was higher in patients with gout than in healthy controls, and this could indicate that constant exposure to UA may predispose monocytes to produce more IL-1β upon stimulation, a theory supported by studies conducted with PBMC22,30. However, despite the trend we observed for increased responsiveness to Pam3 + MSU in monocytes from patients with gout, this was not reflected by increases in the levels of gene expression of components of the NLRP3 inflammasome, nor did these levels correlate with serum UA concentrations. These data suggest that there are sufficient levels of NLRP3 inflammasome components at the protein level to allow secretion of IL-1β in the absence of upregulated gene expression. Significantly, it also suggests that exposure of monocytes to UA does not result in increased gene expression, perhaps explaining why hyperuricemia alone is insufficient to cause IL-1β-mediated inflammation.
Increased expression of NLRP3, PYCARD, CASP1, and IL-1β does occur in conditions such as atherosclerosis31, Sjögren syndrome32, and preeclampsia33, so it is intriguing that we found no increases in basal expression of these genes in patients with gout, given the importance of IL-1β in the disease process in gout. Of note, expression of NLRP3 was relatively low compared to both IL-1β and CASP1, consistent with the requirement for TLR-induced transcriptional priming and translation of NLRP3 prior to oligomerization and activation of the NLRP3 inflammasome15.
The increased ROS in response to UA in the monocytes studied here was intriguing, given the well-known antioxidant effects of UA34. However, UA is also known to have pro-oxidant effects depending on context and location, functioning as a pro-oxidant in intracellular spaces and as an antioxidant in extracellular environments35. This raises the question whether UA enters the monocytes over the course of the incubations or whether UA is exerting these effects on ROS by influencing the extracellular redox environment. Longer incubations with UA did not result in increased ROS in the cells, and this could have been due to increases in cellular antioxidant activity as a compensatory measure. It is unclear whether UA can enter monocytes. UA is taken up by adipocytes36 and vascular smooth muscle cells37 by specific urate transporters, and UA entry into these cell types results in increased ROS. Leukocytes do express the Glut9a urate transporter38 that is associated with regulation of plasma urate levels, but there are no data on whether Glut9a actually transports UA in these cells.
The fact that we see increased ROS in monocytes upon incubation with UA, together with the epigenetic changes that occur in monocytes incubated with UA22, could point to intracellular effects of UA in these cells. However, both these observed effects could also be mediated by UA-induced changes in the extracellular environment that causes changes in the redox state of membrane receptors/proteins that then exert intracellular effects.
The increases in ROS observed after incubation with UA did not influence IL-1β secretion, suggesting that ROS are dispensable for activation of the NLRP3 inflammasome. The role of ROS in IL-1β production is controversial, with some studies showing that activation of the NLRP3 inflammasome is reliant on ROS39,40, but others showing that ROS are dispensable in this process19,41,42. Thus, our findings agree with the view that ROS are not involved in IL-1β production in monocytes.
Our results are consistent with reports that incubation of primary human monocytes with UA did not result in IL-1β secretion23. We found no effect of UA on subsequent stimulation of cells with Pam3 ± MSU, as shown by others23, because we performed these experiments only in THP-1 cells. The mechanism by which preincubation with UA increases IL-1β secretion upon subsequent stimulation with Pam3 is by downregulation of IL-1 receptor antagonist (IL-1Ra), which allows increased binding of the IL-1β produced to the IL-1R22. Crucially, THP-1 cells do not express the IL-1R43 and so cannot respond to IL-1β in this manner. Thus, any downregulation of IL-1Ra in response to UA had no effect in our assays.
What then is the relationship between hyperuricemia and susceptibility to gout? There was no correlation between serum UA and production of IL-1β in monocytes from patients with gout, and our results also appear to show that crystallization of UA into MSU is necessary for activating the NLRP3 inflammasome in these cells. One possibility is that hyperuricemia results in higher levels of oxidative stress where there are greater levels of ROS than can be dealt with by antioxidant mechanisms. The relationship between oxidative stress and inflammation is complex, but these 2 states tend to potentiate each other44,45. It may be that there are less-efficient antioxidant mechanisms operating in patients with gout than in asymptomatic hyperuricemic individuals, resulting in higher levels of oxidative stress that could contribute to greater sensitivity to MSU crystals. Because the presence of MSU crystals within joints does not always result in a gout attack, it may be that a second trigger, perhaps increased oxidative stress, is required to activate the NLRP3 inflammatory cascade. Indeed, this may also explain the association between gout and metabolic syndrome, which is also characterized by increased oxidative stress and increased inflammation46.
ONLINE SUPPLEMENT
Supplementary material accompanies the online version of this article.
Acknowledgment
We are grateful to the clinical research team at the Clinical Investigation and Research Unit, Royal Sussex County Hospital, Brighton, UK, for collection of patient blood samples.
Footnotes
Supported by Brighton and Sussex Medical School.
- Accepted for publication December 20, 2018.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}